


1. Background
Growth is defined as the increase in the size of a tissue or organism, and growth hormone (GH) or somatotropin is an important hormone that regulates the general growth in the human body. GH is synthesized by acidophilic or somatotropic cells of the anterior pituitary gland. Human growth hormone (hGH) has a molecular mass of 22 KDa and contains 191 amino acid residues with two disulfide bridges (1). This hormone involves many biological activities, such as protein synthesis, cell proliferation, protein metabolisms, carbohydrates, and lipids. hGH is in high demand and has been used in clinics since 1985 to treat various children and adult hGH-related disorders, including Prader-Willi syndrome, chronic renal insufficiency, Turner syndrome, and AIDS-related wasting (2).
Genetic engineering provides an exciting approach for heterologous protein production in microbes, especially Escherichia coli. Production of recombinant proteins in bacteria is possible in two ways: One is the cloning of the desired gene in an expression plasmid, and the other is the chromosomal integration of the gene. Hitherto, extensive studies and experiments have been performed on plasmid-based growth hormone production with mixed results depending on the experimental conditions. Recombinant plasmid permanence in the cytoplasm of the host bacteria is associated with challenges such as an increase in the metabolic load of the cell due to the presence of genes and other unwanted parts of the plasmid, the instability of the plasmid, and the need for the constant presence of antibiotics in the bacterial culture media. These problems result in limitations and cost increases in the production process (3).
In the past years, significant progress has been made in recombinant hGH (rhGH) production (4). Zamani et al. optimized the cultivation medium of rhGH in recombinant E. coli through response surface methodology, obtaining an hGH yield of 391 mg/L (5). However, a major hurdle was the formation of protein inclusion bodies (IB) in the host E. coli. Therefore, an efficient recombinant protein expression system is a system where the product is soluble. As a result, the production and downstream processing of the final product is low-cost (6, 7).
Fh8 can effectively increase protein solubility (8) and purification (9). The production of recombinant proteins in E. coli using Fh8 has made the products extremely soluble and converted them to unusually thermostable proteins, which can maintain the integrity of their secondary structures up to 74°C (10, 11).
Integrating the desired heterologous gene fused to the Fh8 tag into the genome may be a valuable strategy to address the mentioned problems for expression in bacterial cells and, perhaps, other hosts. Therefore, the authors decided to integrate engineered DNA cassettes containing the hGH -encoding gene promoter fused to the Fh8 tag into the bacterial chromosome to allow continuous copy number control and reduce the metabolic burden on the host.
In other words, the araBAD operon was considered for inserting the gene cassette into the E. coli chromosome. Then, hGH expression was investigated under the regulation of the operon promoter using the arabinose inducer. In the next step, the DNA cassettes were put under the control of the T7 promoter, and the expression of the recombinant protein was investigated using the IPTG inducer. Finally, the plasmid-based expressions of the DNA cassettes were also investigated, and the results were compared. Considering the unique features of the Fh8 tag, such as improving the increasing the solubility and thermostability of the fused protein’s solubility and increasing the attached protein’s heat resistance, this tag was used in this project to facilitate the research process (12, 13).
2. Objectives
We investigated a method to express human growth hormone stably. We were also looking for a strategy or approach to increase protein production by reducing IB production and facilitating the purification process to form more active and stable proteins.
3. Methods
3.1. Bacterial Strains, Growth Conditions, Plasmids, and DNA Manipulations
The E. coli DH5α (Novagen) was used as a host for cloning and plasmid proliferation. Also, Rosetta-gami and BL21 (DE3) strains were used to integrate the related gene into the E. coli chromosome under the arabinose and T7 promoters, respectively. The plasmid pET28a (+) (Novagen) with kanamycin resistance was used for the plasmid-based expression of the human growth hormone and was assessed in the Rosetta-gami strain. A plasmid without the hGH gene was used as a negative control. The Taq and Pfu DNA polymerase, restriction enzymes NcoI and XhoI, and DNA T4 ligases were obtained from the Fermentas Company. All general chemicals were purchased from Merck. Standard methods were used throughout cloning and manipulating DNA fragments (14). All bacteria were cultured in LB medium supplemented with kanamycin to a final concentration of OD: 0.6 - 0.9.
3.2. Primers, Synthesis of Gene Fragments, and Genetic Manipulations
The GenBank at the NCBI website was used to find the nucleotide and protein sequences of the GH1 gene (IA66930), the E. coli chromosome (arabinose operon) (AP009048), and the Fh8 fragment (AF213970). DNA sequences were evaluated to find the cleavage sites and primer design using software such as the OligoAnalyzer Tool. The primers were synthesized by the Pishgam Company.
3.3. Synthesis of the Fh8-His Tag and Flanking Homology with the Human Growth Hormone
The human growth hormone sequence gene and Fh8 fragment sequence were synthesized by Eurofin after codon optimization for expression in E. coli. The sequence mentioned for the synthesis was without any difference compared to the product of the prominent manufacturing companies.
Fragment 1 included the NcoI recognition sequence and coding sequences for Fh8‚ His tag and the enterokinase cleavage site. The NcoI cleavage site was designed at the 5’ end of the Fh8 (210 bp), which contained the ATG start codon. The His tag (18 bp) was incorporated at the 3’ end of Fh8. Then, the enterokinase cleavage site (15 bp) was designed at the 3’ end of His tag. The total length of the fragment 1 was 250 bp (Figure 1A).
 The schematic of fragment Fh8- His tag and restriction site. (B) The schematic of the hGH fragment and restriction site. (C) The schematic of SOE PCR between fragments 1 and 2.")
(A) The schematic of fragment Fh8- His tag and restriction site. (B) The schematic of the hGH fragment and restriction site. (C) The schematic of SOE PCR between fragments 1 and 2.
Fragment 1, shown in Figure 1, was designed as four pieces, each with almost an 80 bp length (S1 - S4). Each of these pieces overlapped homology with the next part. These fragments were assembled using the polymerase chain reaction (PCR). Fragment 2 (hGH) was amplified using the primers shown in Table 1. The 5’ end of this fragment had homology with the 3’ end of fragment 1, and the XhoI cleavage site was designed at the 3’ end of fragment 2. The length of fragment 2 was 616 bp (Figure 1B). Finally, fragments 1 and 2 were connected by splicing overlap extension PCR (SOEing- PCR), and an end product with an 865 bp length was obtained.
| Name | Sequence (5’ – 3’) | Restriction Site | Function |
|---|---|---|---|
| P.His.Ent.hg | CACCACCATCATGATGACGACGATAAATTTCCGAC | - | Amplification of rhGh |
| Rrhgh-p32 | AGGTCTCGAGATTAAAAGCCACAACTCC | XhoI | |
| FfhS1 | GATCACCATGGCACCGTCTGTTCAGGAAGTTGAGAAACTGCTGCACGTTCTGGACCGTAATGGTGATGGTAAAGTTTCTGCGGAAG | NcoI | Construction of FH8-his tag with homology with rhGh length: 250 bp |
| RfhS2 | TTGTCGTGTTCTTTGATGAACGCTTTGATCTTGTTAGAGTCCAGCGGGCATTTAGAGTCGTCCGCGAACGCTTTCAGTTCTTCCGCAGAAACTTTACCATCACCAT | - | |
| FfhS | TGCCCGCTGGACTCTAACAAGATCAAAGCGTTCATCAAAGAACACGACAAGAACAAAGACGGTAAACTGGACCTGAAAGAACTGG | - | |
| RfhS4 | TTTATCGTCGTCATCATGATGGTGGTGaTGaTGAGAAGACAGGATAGAAACCAGTTCTTTCAGGTCCAGTTTACCGTCTTTG | - | |
| 50araUT7p | GCAACTCTCTACTGTTTCTCCATACCCGTTTTTTTGGATGGAGTGAAACGCGATCCCGCGAAATTAATACGACTCACTATAGG | - | Amplification of rhGH with T7 promoter |
| Rrhgh-p32 | AGGTCTCGAGATTAAAAGCCACAACTCC | XhoI |
A pair of primers of flanking homology with arabinose operon were designed for insertion into the E. coli chromosome. The forward primer had homology with the arabinose promoter in 50 nucleotides of (70049 - 70098), and the reverse primer had homology with the arabinose terminator in 50 nucleotides of (65780 - 65830) (Figure 1C). Moreover, to insert the growth hormone gene into the E. coli genome, the hGH gene in the pET28 vector was used as a template in a manner that one primer was designed for the upstream of the T7 promoter, and the other one was designed for the upstream of the kanamycin resistance promoter. These genes are coded in different strands in the PET28 vector; therefore, the T7 promoter was placed at one end, and the kanamycin resistance gene promoter was placed at the other end of the fragment. Then these two primers were used to amplify the fragment. The two primers had about 50 nucleotides of homology with the arabinose operon. Thus, after transforming the PCR product into a competent cell with an induced λ Red recombination system, kanamycin-resistant colonies peaked and were assessed for the presence of the hGH gene in their genome (15).
3.4. Electroporation, Integration of Gene Cassette in the Escherichia coli Chromosome
The cassette made for integration into the genome was transformed by electroporation into a competent E. coli, which had already received the plasmid encoding lambda RED at 37°C, 180 rpm for three hours. This bacterium was induced with a final concentration of 0.2% arabinose to activate the RED recombining system. After the bacteria of the previous stage grew and reached OD = 0.5, it was washed three times with ultrapure deionized water. Forty microliters of these cells were mixed with one microliter of cassette and transformed by a GenePulseII electroporator with 25 microfarad power, 200 Ohm resistance, and 1.8 kV.
After adding SOC culture medium to the bacteria and incubation at 30 degrees Celsius for two hours, they were spread on a Luria-Bertani (LB) culture medium containing kanamycin. After overnight growth, the plates were examined, and the grown colonies were further examined in the following steps.
The recombinant plasmid containing the rhGH gene was transformed into the bacteria using the conventional heat shock method and CaCl2 solution.
The correct integration of the recombinant cassette at the appropriate position in the bacterial genome was confirmed using two pairs of primers. One pair was for the upstream of the arabinose operon and the integrated cassette in the genome, and another pair of primers was designed to amplify the end of the integrated fragment. The final confirmation was done by sequencing these PCR products.
3.5. Expression of Recombinant Human Growth Hormone
After confirmation of the constructed cassette by PCR, the recombinant colonies were cultured in 5 mL of LB at 37°C until OD = 0.9. These cultures were used as inoculum for 50 mL of fresh LB. After growing to OD = 0.6 - 0.9, they were induced with IPTG or arabinose with final concentrations of 0.5 mM and 0.2%, respectively.
To check the expression in bacteria, sampling was done at the induction time and 4, 8, 15, and 20 hours afterward.
3.6. Sonication, SDS-PAGE, and Western Blot
The cell lysis buffer was prepared (Tris 20 mM, Triton X100 0.5%, EDTA 1 mM) and autoclaved based on the standard protocol. The samples were sonicated on ice for 5 minutes at 10-second bursts at 200W with 10-second cooling down intervals on ice between each burst. Following sonication, standard SDS-PAGE gels (12 and 14.5% acrylamide) were used according to the Laemmlei protocol (16) to evaluate the recombinant protein expression. Also, the expressed rhGH was confirmed by Western blot. The primary antibody was the rabbit monoclonal antibody against hGH (1:500 dilution), and the secondary antibody was anti-rabbit IgG (1:10,000 dilution).
3.7. Purification of Fh8-hGH Fusion Protein
Recombinant bacteria were cultured and induced according to the previous conditions. Then 250 milliliters of the 18-hour culture were centrifuged at 4000 rpm, and the pellet was kept in a -20 freezer. The pellets were removed from the freezer and placed on ice for a few minutes, then 40 milliliters of lysis buffer was added (50 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.1% Triton X-100, 1 mg/mL lysozyme, 1 × protease inhibitor cocktail with the addition of 10 mM imidazole after centrifugation) to purify the protein. Next, sonication was performed for 5 minutes under the same conditions with 10-second bursts at 200W and 10 seconds cooling down intervals on ice between each burst. Afterward, the lysate was centrifuged at 10,000 rpm for 20 minutes. The resulting supernatant was transferred to another vial and heated at 68°C for 15 minutes. At this stage, the supernatant was separated (centrifuged at 22000 rpm for 20 minutes at 4°C) and purified in a Ni Sepharose column. For this purpose, a 2 mL Ni Sepharose 6 FF column (Cytiva, Uppsala, Sweden) was used with a 0.3 mL/min flow rate. The column was washed with 3 column volumes of washing buffer (1× PBS, pH 7.4, containing 150 mM NaCl, 20 mM imidazole, 0.1% Triton X-100, and 10% glycerol). Fh8-hGH was eluted with two column volumes of elution buffer (1× PBS, pH 7.4, containing 150 mM NaCl, 300 mM imidazole, 0.1% Triton X-100, 10% glycerol) in 1 mL fractions at the flow rate 0.5 mL/min. The protein purity of the elution fractions was monitored by 14% SDS-PAGE.
4. Results
4.1. Investigation of the Genomic Expression of Fh8-hGH
4.1.1. Integration of the Fh8-hGH Cassette Into the Rosetta-gami Genome
Synthesis of the Fh8-hGH fragment: First, the Fh8-His tag fragment was designed, assembled, and amplified. The hGH gene was amplified using the primers given in Table 1. Then, the two fragments were joined to form Fh8-hGH using the SOE-PCR.
Cloning of the Fh8-hGH into an expression vector: The Fh8-hGH cassette and the vectors pET28a were digested using the NcoI and XhoI enzymes, then the cassette was ligated to the vector using DNA ligase. After PCRs (Table 2) confirmation and sequencing, the recombinant plasmid was introduced into the Rosetta-gami strain.
| Name | Sequences (5’ - 3’) | Confirmation Gene Fragment Length |
|---|---|---|
| F500araU | CGGGTATGGAGAAACAGTAGAGAGTTG | 2200 bp |
| S4 | TTTATCGTCGTCATCATGATGGTGGTGaTGaTGAGAAGACAGGATAGAAACCAGTTCTTTCAGGTCCAGTTTACCGTCTTTG | |
| F500ara U | CGGGTATGGAGAAACAGTAGAGAGTTG | 2800 bp |
| Rgh32 | AGGTCTCGAGATTAAAAGCCACAACTCC | |
| S1 | GATCACCATGGCACCGTCTGTTCAGGAAGTTGAGAAACTGCTGCACGTTCTGGACCGTAATGGTGATGGTAAAGTTTCTGCGGAAG | 1550 bp |
| R500araD | GCCAATCAAACGAAACCAGGCTATAATCAAGCCTGG | |
| FNcoI | GATCACCATGGCACCGTCTGTTCAGGAAG | 1000 bp |
| R500araD | GCCAATCAAACGAAACCAGGCTATAATCAAGCCTGG |
Integration of Fh8-hGH cassette into the Rosetta-gami genome: The homologous recombination by the λ-Red system was used to integrate the cassette into the E. coli chromosome. For this purpose, the plasmid pKD46 was transformed into the Rosetta-gami strain by electroporation. The recombinant plasmid pET28-Fh8-hGH was used as a template to amplify the Fh8-hGH cassette, which was flanked with 50 nucleotide homologs sequences with the promoter and terminator of the arabinose operon at 3’ and 5’ end, respectively. At the same time, the reverse primer was designed at the beginning of the kanamycin resistance gene in the pET28 vector. The PCR-amplified fragment was 2363 bp, including the kanamycin marker gene, Fh8-hGH, and the nucleotide sequence between them.
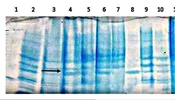
This cassette was cleaned, dissolved in double distilled deionized water, and transformed into competent recombining bacteria by electroporation. The transformants were cultured on LB agar containing 15 μg/mL kanamycin. After overnight incubation at 30°C, single colonies were confirmed by PCR (Figure 2A). Confirmed colonies were induced with a final concentration of 0.2% arabinose, and their expression was evaluated by SDS-PAGE (Figure 3).
 1 - 6: 1000 bp fragment of recombinant hGH integrated into Rosetta-gami (DE3) chromosome by FNcoI and R500araD primers, 7: Ladder. (B) 2 - 4: 1550 bp fragment of recombinant hGH integrated into the BL21 (DE3) chromosome by S1 and R500araD primers, 1,5: Ladder.")
Confirmation of the insertion of the recombinant cassettes in the correct position of the arabinose operon by PCR. (A) 1 - 6: 1000 bp fragment of recombinant hGH integrated into Rosetta-gami (DE3) chromosome by FNcoI and R500araD primers, 7: Ladder. (B) 2 - 4: 1550 bp fragment of recombinant hGH integrated into the BL21 (DE3) chromosome by S1 and R500araD primers, 1,5: Ladder.
 chromosome under the arabinose promoter. (1) protein ladder. (2) T<sub>20</sub> Colony 6. (3) T<sub>4</sub> Colony 6. (4) T<sub>20</sub> Colony 7. (5) T<sub>4</sub> Colony 7. (8) Positive control. (11) Negative control. (6, 7, 9, 10, 12, 13, 14, 15, 16, 17) Study colonies.")
Expression of the recombinant hGH integrated into Rosetta-gami (DE3) chromosome under the arabinose promoter. (1) protein ladder. (2) T20 Colony 6. (3) T4 Colony 6. (4) T20 Colony 7. (5) T4 Colony 7. (8) Positive control. (11) Negative control. (6, 7, 9, 10, 12, 13, 14, 15, 16, 17) Study colonies.
4.1.2. Integration of the T7 Promoter/Fh8-hGH Cassette Into the BL21 (DE3) Genome
Synthesis of the cassette containing the T7 promoter- This stage included adding the T7 promoter upstream of the hGH gene. The forward primer binding sequence was designed upstream of the T7 promoter of pET28-Fh8-hGH to amplify the cassette. The cassette was amplified with forward primer 50 araut7p and reverse primer 50araR. The preparation of recombineering competent cells of E. coli BL21 (DE3) and transformation of the cassette were done in the same way as the previous steps. The only difference was in the induction of transformants. In this step, IPTG with a concentration of 1 μM was used for induction. Single colonies were confirmed by PCR (Figure 2B).
The assessments were performed 4 and 16 hours following the induction, and the results were compared (Figure 4).
 chromosome under the control of the T7 promoter after 20 hours of induction. (1, 2, 3, 4, 5, 6, 8, 9, 10, 12, 13) study colonies, (7) positive control, (11) negative control, and (14) marker.")
Expression of the recombinant hGH integrated into the BL21 (DE3) chromosome under the control of the T7 promoter after 20 hours of induction. (1, 2, 3, 4, 5, 6, 8, 9, 10, 12, 13) study colonies, (7) positive control, (11) negative control, and (14) marker.
4.2. Evaluation of the Expression from the Recombinant Plasmid pET28-Fh8-hGH in Rosetta-gami
Recombinant plasmid pET28-Fh8-hGH was transformed into the E. coli Rosetta-gami, and the plasmid-based expression was assessed after induction with IPTG 1 µM (Figure 5).
 T4 of study colonies, (3) negative control, (4) positive control, and (5) marker.")
Episomal expression of recombinant hGH, pET28-a Fh8-hGH in Rosetta-gami, (1, 2) T4 of study colonies, (3) negative control, (4) positive control, and (5) marker.
4.3. Western Blot
The recombinant protein expressed in the recombinant strains of E. coli, which were used in this research, after initial confirmation by SDS-PAGE, was confirmed by western blot using anti-hGH monoclonal antibody (Figure 6).
 <i>Escherichia coli</i> Rosetta-gami P.ara (Fh8-rhGH), (2) <i>E. coli</i> BL21 (DE3) P.T7 (Fh8-rhGH), (3) Marker, (4) positive control, and (5) <i>E.coli</i> Rosetta-gami pET28-Fh8-rhGH.")
Western blotting of the Fh8-rhGH produced by recombinant strains. (1) Escherichia coli Rosetta-gami P.ara (Fh8-rhGH), (2) E. coli BL21 (DE3) P.T7 (Fh8-rhGH), (3) Marker, (4) positive control, and (5) E.coli Rosetta-gami pET28-Fh8-rhGH.
4.4. Effect of Fh8 on Expression Level, Solubility, and Temperature Stability
For this purpose, after culturing the recombinant strains and their sonication, the cell supernatant was heated at 68 degrees for 20 minutes and compared with the non-heated supernatant (Figure 7 and 8).

Comparison of recombinant fusion protein expression in soluble and sediment fractions in recombinant Escherichia coli strain pET28-Fh8-rhGH after sonication and heating in 68°C. Lanes 1, 3, 5: Soluble fractions of colonies. Lanes 2, 4: Sediment fraction of colonies.
 SDS-PAGE of the supernatant fraction of cell lysate after sonication without heat treatment (1) <i>Escherichia coli</i> BL21 (DE3) P.T7 (Fh8-rhGH), (2) Marker and (B) with heat treatment at 68°C. (1, 2, 3) <i>E. coli</i> BL21 DE3) P.T7 (Fh8-rhGH) and (4) marker.")
(A) SDS-PAGE of the supernatant fraction of cell lysate after sonication without heat treatment (1) Escherichia coli BL21 (DE3) P.T7 (Fh8-rhGH), (2) Marker and (B) with heat treatment at 68°C. (1, 2, 3) E. coli BL21 DE3) P.T7 (Fh8-rhGH) and (4) marker.
4.5. Purification of Fh8-hgh Fusion Protein
Rosetta-gami (DE3) cells expressing recombinant hGH (Fh8-(His)6-hGH) cassette in its genome were grown in 1 L flasks containing 250 mL of optimized culture medium and induced at 37°C for 18 hours and shaking at 250 rpm. Harvested cells (≈ 2.5 g) were suspended in 25 mL of lysis buffer (50 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.1% Triton X-100, 1 mg/mL lysozyme, 1× protease inhibitor cocktail with the addition of 10 mM imidazole after centrifugation), disrupted by sonication, and then centrifuged at 10,000 g for 15 minutes. To remove the maximum number of unwanted proteins and reduce the variety of proteins, heat treatment was performed at 68 degrees Celsius for 20 minutes. To remove the inherently insoluble proteins or those denatured due to heat becoming insoluble, the samples were centrifuged at 22000 rpm and 4°C for 20 minutes. Ten mM of imidazole was then added to the supernatant. The supernatant was applied onto a 2 mL Ni Sepharose 6 FF column (Cytiva, Uppsala, Sweden) with 0.3 mL/min flow rate to purify the Fh8-(His)6-hGH. The column was then washed with 3-column volumes of washing buffer and eluted with 2-column volumes of elution buffer in 1 mL fractions at the flow rate of 0.5 mL/min. The protein purity of the elution fractions was monitored by SDS-PAGE (Figure 9).
 from <i>Escherichia coli</i> BL21 (DE3) P.T7 (Fh8-rhGH). (1, 2) The results obtained from the 3 mL fraction. (3) Marker.")
Purified recombinant hGH (Fh8-hGH) from Escherichia coli BL21 (DE3) P.T7 (Fh8-rhGH). (1, 2) The results obtained from the 3 mL fraction. (3) Marker.
5. Discussion
The most common and successful expression method of recombinant proteins is episomal expression. However, this method has some apparent weaknesses. The metabolic load caused by the plasmid can lead to the reduction of the host bacteria’s growth and instability of the plasmid encoding the recombinant gene (17).
On the other hand, overexpression under strong promoters such as T7 may, in many cases, lead to the formation of IBs that are sometimes difficult to refold. Although these intracellular bodies may protect the heterologous protein from the damage of cellular proteases and are also useful in the initial stages of protein purification, refolding some proteins is a serious challenge. This can increase the cost of the final product (18) and is documented in the case of human growth hormone. When this hormone was expressed in E. coli at 37°C, almost all the expressed protein was in the form of IB (19). Therefore, reducing or preventing the formation of IBs can facilitate the purification process of some proteins, including human growth hormones.
The instability of the recombinant plasmid and the metabolic load caused by the high copy number of the plasmid may be solved or reduced by integrating the heterologous gene in the appropriate position of the host genome (20). The Lambda red recombination system is an efficient tool for gene integration in the E. coli genome (15), and we successfully applied this recombineering system to integrate the target gene, rhGH, into the E. coli genome.
To achieve stable expression, possibly facilitate purification, and ultimately reduce the cost of recombinant human hormone (rhGH) production, we inserted this gene along with a tag with unique characteristics, Fh8, into the E. coli Bl21 genome. In this manner, the plasmid curing problem in the absence or reduction of an effective amount of antibiotics during the next stages of the cultivation of recombinant bacteria was solved. In addition, due to the coordination of the rhGH gene with genome replication, the risk of mutation in this gene was also decreased.
Since removing the arabinose operon does not damage E. coli, the araBAD operon was used for integration, and as shown in Figure 6, an acceptable expression was observed in these strains. However, this amount of expression needed to be increased. Considering that the T7 promoter is one of the most powerful promoters for gene expression (21), it was decided to use this promoter upstream of the integrated gene. Of course, genomic expression was expected to be lower than plasmid expression under the same promoter due to the difference in copy numbers. To investigate whether this weakness can be partially compensated using two promoters, a cassette containing the human growth hormone coding gene with T7 promoter in its 5’ end was integrated downstream of the arabinose promoter (i.e., araBAD). In this case, it was possible to induce one of the two promoters or both promoters together.
Previous studies have not reported a significant difference in the simultaneous induction of both promoters (22), but the amount of expression was slightly higher than the individual induction of the promoters; therefore, simultaneous induction was employed in this study.
Costa reported that the presence of the Fh8 tag, together with a recombinant green fluorescent protein (GFP) and the superoxide dismutase (SOD) as target model proteins, improved the expression of the protein (13). Accordingly, the decision to add the Fh8 tag coding sequence to the rhGH gene was made to increase expression in this study. In addition, the presence of the Fh8 tag, along with the rhGH gene, could increase the level of solubility due to its fused protein preventing or reducing the formation of inclusion bodies. Additionally, this tag gives a high thermal stability to the protein, which maintains the second structure of the protein up to 70°C (12). This feature can also be used for easier and faster protein purification.
Cultivation and induction were repeated after confirming the expressed protein by Western blotting using anti-hGH monoclonal antibodies, as shown in Figure 6. The bacteria were then lysed using ultrasound, and the pellet and supernatant were separated and analyzed by SDS-PAGE. As shown in Figure 7, a significant fraction (69.86%) of the protein was expressed as soluble, and a smaller fraction (30.14%) as insoluble.
Since Fh8 and human growth hormone can tolerate high temperatures, this characteristic was used to remove some host proteins in the purification stage (12, 23). The supernatant of samples was heated at 68°C for 20 minutes, denatured most of the proteins. Insoluble denatured proteins were removed by a high-speed centrifuge. In Figure 8, the comparison between the supernatants before and after heating with SDS-PAGE indicated eliminating some protein bands, but the recombinant protein band did not change.
Lastly, the His-tag was used for simple and highly selective product purification of the product (10). Therefore, a Nickle Sepharose column was used as the last purification step, as shown in Figure 9, and the relatively pure Fh8-rhGH was obtained.
5.1. Conclusions
A comparative study of genomic and episomal expression demonstrated that although genomic expression is less than episomal expression, each cell had 15 - 60 copies of pET plasmids. If this amount of expression is divided for each copy of the gene in the cell, the expression of a genomic copy will be higher than the expression of a single plasmid copy. Therefore, it appears that integrating more copies of the desired gene can facilitate stable yet higher expression achievement compared to its episomal form. In addition, it should be considered that the high expression of a recombinant protein, even from the integrated versions in the genome, may be confronted with the critical challenge of inclusion body formation. This challenge can be overcome by fusing a suitable tag, such as Fh8, to the desired gene and then integrating it into appropriate positions in the genome.
