



1. Background
Currently, maintaining a clean environment has become one of the essential tasks of humankind. In general, the inattention to pollution particles of the environment and living organisms is naturally followed by numerous environmental problems, diseases, and cancers. A part of these pollution particles exists in water, such as toxic and heavy metals (metal ions) in water and/or wastewater. The increasing rate of environmental problems in numerous societies has made it necessary for research groups to put forward new and logical solutions for this issue. Therefore, the current study focused on proposing a new method to remove heavy metal ions (HMIs) from water and/or wastewater.
The removal of HMIs as one of the pollution sources in seawater or industrial wastewater needs to adsorb on large surface areas and high active sites (1-20). Therefore, nanostructures with these properties are introduced as excellent candidates (1, 21). For example, nanostructures with a good ability of adsorption were introduced to remove heavy metal atoms (1). In another theoretical study, graphene-based materials were introduced for the adsorptive removal of pollutants, such as HMIs, from water (21). For instance, amine-functionalized graphene oxide was used as a sorbent to achieve carbon dioxide capture (21). Therefore, theoretical studies can help to the precise understanding of the interaction between nanostructures and HMIs. This theoretical study proposes logical methods to experimental research groups for the removal of pollution particles.
2. Objectives
The existence of HMIs (zinc(II) ion [Zn2+]/cadmium(II) ion [Cd2+]/mercury(II) ion [Hg2+]) in seawater or wastewater results in numerous environmental problems, diseases, and cancers. Therefore, the natural removal of HMIs is followed by decreasing the aforementioned problems. The major objective of this study was to investigate the adsorption of HMIs (Zn2+/Cd2+/Hg2+) in zigzag nanosheets (NSs), such as functionalized carbon nanosheet (CNS) {(NH2)n-CNS (n = 1 - 2)}, using density-functional theory (DFT). The HMIs are susceptible to binding to {(NH2)n-CNS (n = 1 - 2)} structures. Therefore, the present study will optimize CNS {(NH2)n-CNS (n = 1 - 2)} structures and their complexes with HMIs (Zn2+/Cd2+/Hg2+) to obtain their ground states. In addition, the quantum theory of atoms in molecules (QTAIM) and natural bond orbital (NBO) analysis confirm the formed bonds and interactions between {(NH2)n-CNS (n = 1 - 2)} structures and HMIs (Zn2+/Cd2+/Hg2+) in the best configuration and orientation.
3. Methods
The methodological approach in this study was based on quantum mechanics studies. The advantage of these methods is gaining insight into CNS and {(NH2)n-CNS (n = 1 - 2)} chemical reactivity and their interactions with HMIs (Zn2+/Cd2+/Hg2+), respectively. The optimization of CNS, {(NH2)n-CNS (n = 1 - 2)}, and HMIs were carried out using a Gaussian software package (version 09) to obtain their low-lying structures (22). In the framework of quantum calculations, hybrid DFT methods, including the wB97XD exchange-correlation (xc) functional (23). with pseudopotential basis sets and lanl2dz, were used (24). Ground state with no imaginary frequencies was evaluated by frequency test. This work confirms that all structures are in actual minima. In addition, the molecular electrostatic potential (MEP) (25). was performed to better understand electronic structures and their role in reactions. For the investigation role of CNS, {(NH2)n-CNS (n = 1 - 2)}, and HMIs (Zn2+/Cd2+/Hg2+) from seawater in interaction, their global and local descriptors were calculated in the DFT-based calculations.
Reactivity is the tendency to undergo a possible chemical reaction with other molecules (26). Global chemical reactivity descriptors are chemical potential (µ=
IE (compond) = Etot (compound+) - Etot (compound)
EA (compound) = Etot (compound-) - Etot (compound)
The active sites of CNS and {(NH2)n-CNS (n = 1 - 2)} for interaction with HMIs (Zn2+/Cd2+/Hg2+) were predicted by the investigation of local descriptors, such as Fukui function (f) and local softness (s). The f is the sensitivity of the chemical potential of a system to a local external potential (26). Finite difference approximation was used to characterize nucleophilic (f+) and electrophilic (f -) attacks as follows: (26)
Where
When α=+,- means nucleophilic, electrophilic attacks, the maximum positive values of local softness indicate most electrophile site of atom in the compound. Therefore, this site is favorable for nucleophilic attacks in reaction with other molecules.
The QTAIM (27) and NBO analysis (28) complete geometric and electronic information of zigzag CNS, {(NH2)n-CNS (n = 1 - 2)}, and HMIs (Zn2+/Cd2+/Hg2+) interactions. Electron density ρ(r) and Laplacian ∇2ρ(r), electronic energy density H(r), electronic kinetic energy density G(r), and electronic potential energy density V(r) descriptors were calculated at bond critical points (BCPs) based on the QTAIM theory. Moreover, the type of bonds, depletion of occupancies, percentage of Lewis and nonLewis, and stabilization energies 28 were obtained by NBO analysis. The second-order stabilization energy (
Where
4. Results and Discussion
Zigzag CNS and {(NH2)n-CNS (n = 1 - 2)} structures (CNS (n, m; L), and L equals to length (in angstrom, Å) are considered substrates for the adsorption and removal of HMIs (Zn2+/Cd2+/Hg2+) from seawater. The zigzag CNS and {(NH2)n-CNS (n = 1 - 2)} have chirality (10, 0) with 10 Å in length properties. The low lying structures of zigzag CNS and {(NH2)n-CNS (n = 1 - 2)} structures were investigated in the wB97/Lanl2dz level of theory (Figure 1). Symmetrical constraints (C1 symmetry) were not considered in optimization processing that leads to the exact investigation of ground states. On the other hand, frequency calculation confirmed this issue because there were no imaginary frequencies.

Optimal ground state structures of nanosheet structures in wB97XD/lanl2dz method
The investigation of active sites and the prediction of the strength of interactions in isolated {(NH2)n-CNS (n = 1 - 2)} structures were carried out using MEP maps (25) (Figure 2). Low and high electron density was related to the positive and negative points of charge distribution in the MEP map, respectively. The yellow, blue, and green colors elucidate the most negative, positive, and zero electrostatic potentials, respectively (Figure 2). According to Figure 2, adding one and/or two NH2 groups mainly results in decreasing positive electrostatic potential in the center of CNS nanostructure. This issue confirms that the functionalization of CNS nanostructure can increase the chemical reactivity of this structure to interact with HMIs (Zn2+/Cd2+/Hg2+).

Calculated molecular electrostatic potential of nanosheet structures using wB97XD/lanl2dz method
The confirmation of the above-mentioned behavior of CNS and {(NH2)n-CNS (n = 1 - 2)} structures is understandable with the investigation of their role in the interaction. A simple way to achieve this purpose is using global chemical reactivity descriptors, as shown in Table 1. High and low negative values of the µ in HMIs (Zn2+/Cd2+/Hg2+) and CNS, {(NH2)n-CNS (n = 1 - 2)}, indicated that they accept and donate electrons conveniently, respectively. Therefore, the HMIs (Zn2+/Cd2+/Hg2+) and CNS, {(NH2)n-CNS (n = 1 - 2)}, NS play electrophile and nucleophile roles in their interaction. This issue is in line with the high η and low S of CNS and {(NH2)n-CNS (n = 1 - 2)} structures. Furthermore, the high ω of nucleophile showed that the floating of the electron was carried out from NSs to HMIs (Zn2+/Cd2+/Hg2+) conveniently.
| Descriptors (eV) | CNS | (NH2)-CNS | (NH2)2-CNS | Zn2+ | Cd2+ | Hg2+ |
|---|---|---|---|---|---|---|
| IE | 3.005 | 2.8148 | 2.8639 | 28.8148 | 32.0089 | 29.9731 |
| EA | 0.5648 | 0.2276 | 0.7892 | 20.4476 | 19.4673 | 20.9037 |
| μ | -1.7851 | -1.5212 | -1.8266 | -24.6312 | -25.7381 | -25.4384 |
| η | 1.2203 | 1.2936 | 1.0373 | 4.1836 | 6.2708 | 4.5347 |
| S | 0.8195 | 0.7731 | 0.9640 | 0.2390 | 0.1595 | 0.2205 |
| ω | 1.3057 | 0.8944 | 1.6081 | 72.5095 | 52.8201 | 71.3510 |
Ionization Energy, Electron Affinity, Chemical Potential (μ), Global Hardness (η), Softness (S), and Electrophilicity (ω) of Carbon Nanosheet, {(NH2)n-CNS (n = 1 - 2)}, and Heavy Metal Ions, Respectively
The local chemical reactivity method can help investigate the exact active sites of the NS in the interaction with HMIs (Zn2+/Cd2+/Hg2+). According to Table 2, the local softness (
| Atoms | f + | f - | s+ | s- |
|---|---|---|---|---|
| C | 0.01651 | -0.01505 | 0.01592 | -0.01450 |
| N | 0.00417 | -0.00396 | 0.00402 | -0.00381 |
Calculated Local Descriptors of {(NH2)n-CNS (n = 1 - 2)} Structures in Selected Level of Theory
For more understanding of {(NH2)n-CNS (n = 1 - 2)} NSs and HMIs (Zn2+/Cd2+/Hg2+) interaction, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) have been plotted (Figure 3). According to Figure 3, the HOMO (NH2)2-CNS-LUMO HMI gap was decreased by about 1.35 eV more than the other NSs. Therefore, there is probably a strong charge transfer in HMIs → (NH2)2-CNS and (NH2)2-CNS → HMIs, respectively.
<sub>n</sub>-CNS (n = 1 - 2)} structures, and heavy metal ions (Zn<sup>2+</sup>/Cd<sup>2+</sup>/Hg<sup>2+</sup>)")
Highest Occupied molecular orbital and lowest unoccupied molecular orbital gaps of carbon nanosheet, {(NH2)n-CNS (n = 1 - 2)} structures, and heavy metal ions (Zn2+/Cd2+/Hg2+)
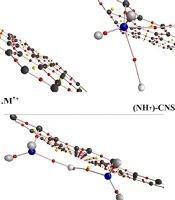
With the determination of the ground state of structures, the HMIs (Zn2+/Cd2+/Hg2+) as ligand interacted with CNS and {(NH2)n-CNS (n = 1 - 2)} structures. The low lying structures of the open and closed-shell ({(NH2)n-CNS (n = 1 - 2)}- M2+ (M2+= Zn2+/Cd2+/Hg2+) complexes were investigated by the selected level of theory (Figure 4). The more negative value of the Eads calculations implies that the (NH2)2-CNS-M2+ complexes have higher adsorption strength and stable configuration than the CNS- M2+ and (NH2)-CNS-M2+ structures, as shown in Table 3. Therefore, the functionalized CNS structures have a tendency to interact with the HMIs. According to Table 3, the adsorption energy value in (NH2)2-CNS-Hg2+ is more than other complexes. This issue can be due to the interaction of two NH2 groups with the HMIs.
<sub>n</sub>-carbon nanosheet (n = 1 - 2)}-M<sup>2+</sup> (M<sup>2+</sup>= Zn<sup>2+</sup>/Cd<sup>2+</sup>/Hg<sup>2+</sup>) complexes at selected level of theory")
Optimized geometry of ({(NH2)n-carbon nanosheet (n = 1 - 2)}-M2+ (M2+= Zn2+/Cd2+/Hg2+) complexes at selected level of theory
| Complexes | Zn | Cd | Hg |
|---|---|---|---|
| CNS | -4.31 | -3.09 | -7.43 |
| CNS (NH2) | -10.27 | -13.34 | -17.43 |
| CNS (NH2)2 | -75.72 | -84.39 | -93.48 |
Calculated Adsorption Energies of Carbon Nanosheet (CNS) and CNS(NH2)n (n = 1 - 2)… M2+ Complexes in wB97XD/lanl2dz Method
4.1. QTAIM and NBO Analysis
The evaluation of the CNS… M2+ and {(NH2)n-CNS (n = 1 - 2)}… M2+ (M2+= Zn2+/Cd2+/Hg2+) interactions was carried out using QTAIM theory (27). Figure 5 depicts the bond paths (BPs) and BCPs of NSs and HMIs (M2+= Zn2+/Cd2+/Hg2+) interactions. According to Figure 5, there is no BP and BCPs in the CNS… M2+ systems. Table 4 shows the calculated descriptors at the BPs and BCPs of these interactions.
- M<sup>2+</sup> and (NH<sub>2</sub>)<sub>x</sub>-CNS- M<sup>2+</sup> (M= Zn<sup>2+</sup>, Cd<sup>2+</sup>, Hg<sup>2+</sup>, x = 1 - 2); Bond critical point (in red) and ring critical point (in yellow)")
Molecular graph of carbon nanosheet (CNS)- M2+ and (NH2)x-CNS- M2+ (M= Zn2+, Cd2+, Hg2+, x = 1 - 2); Bond critical point (in red) and ring critical point (in yellow)
| Complexes, Bond | ρ(r) | ∇2ρ(r) | H(r) |
|---|---|---|---|
| (NH2)-CNS- Zn2+ | |||
| C-N | 0.2378 | 0.10160 | 0.197717 |
| N-Zn2+ | 0.01816 | -0.01313 | 0.000449 |
| (NH2)-CNS-Cd2+ | |||
| C-N | 0.1704 | 0.12960 | 0.176100 |
| N-Cd2+ | 0.0171 | -0.01498 | 0.000762 |
| (NH2)-CNS-Hg2+ | |||
| C-N | 0.2181 | 0.10260 | 0.16043 |
| N-Hg2+ | 0.01795 | -0.01342 | 0.000726 |
| (NH2)2-CNS-Zn2+ | |||
| C-N left | 0.2252 | 0.08507 | 0.21998 |
| C-N right | 0.2346 | 0.09714 | 0.20867 |
| N-Zn2+ left | 0.08971 | -0.10277 | 0.023108 |
| N-Zn2+ right | 0.09731 | -0.11368 | 0.026925 |
| (NH2)2-CNS-Cd2+ | |||
| C-N | 0.1159 | 0.09370 | 0.23007 |
| C-N | 0.1250 | 0.1027 | 0.21803 |
| N-Cd2+ | 0.08243 | -0.10781 | 0.03169 |
| N-Cd2+ | 0.07510 | -0.11627 | 0.031015 |
| (NH2)2-CNS-Hg2+ | |||
| C-N | 0.13261 | 0.09104 | 0.16181 |
| C-N | 0.13038 | 0.09539 | 0.15882 |
| N-Hg2+ | 0.09224 | -0.11020 | 0.04021 |
| N-Hg2+ | 0.09268 | -0.12053 | 0.04537 |
Analysis of Bond Critical Points in Covalence and Electrostatic Bonds at (NH2)n-CNS… M2+ (n = 1 - 2) Complexes
Probably the interactions in the CNS… M2+ systems are very weak. This issue is in line with Eads calculations. Additionally, the information of the BCPs in the {(NH2)n-CNS (n = 1 - 2)}… M2+ confirms the interaction between functionalized NSs and HMIs. Therefore, the electron density values of ρ(r) in the BCP of this interaction were observed lower than the covalence bonds (Table 4). Moreover, the ρ(r) values decreased with an ascending trend in the atomic mass of HMIs and lowering distances in the 2HN…. M2+ interactions. This issue is in line with the elongation of the C–N bond. Therefore, high ρ(r), more elongation of the C–N bond, low 2HN… M2+ distance, and linearity of the M2+…N-C angle lead to the charge transfer of nitrogen atom to HMIs. The small values of the ∇2ρ(r) and H(r) in 2HN…. M2+ indicated that the nature of these bonds is electrostatic. The positive and negative values of the ∇2ρ(r) in C–N, and 2HN…. M2+ bonds implied that these bonds have covalence and electrostatic natures. The high and low values of the H(r) descriptor in these bonds signified that the nature of such bonds was decreased and increased.
In spite of the fact that the QTAIM has high performance to investigate these types of interactions, it cannot identify the source of changes and charge transfers (27). Therefore, NBO analysis (28) showed that there are two significant donor-acceptor interactions in the {(NH2)n-CNS (n = 1 - 2)}… M2+ systems, such as nN→ lp*M2+ and lpM2+→ σ*C–N, with the second-order stabilization energies of 65.8 and 13.01 kcal mol-1, respectively. Consequently, nN→ lp*M2+ interaction was stronger than lpM2+→ σ*C–N, which is in line with QTAIM analysis.
5. Conclusions
The CNS structures can help to the removal of HMIs (Zn2+/Cd2+/Hg2+) from water, especially seawater and/or wastewater. For the achievement of this purpose, CNS and amine-functionalized NS ({(NH2)n-CNS (n = 1 - 2)}) interacted with HMIs (Zn2+/Cd2+/Hg2+). Based on DFT, (NH2)2-CNS has a good ability for the removal of HMIs due to high chemical potential (-1.827 eV), softness (0.964 eV), high electrophilicity (1.608 eV), and adsorption energy (≈ -75 to -93 kJ mol-1) in the (NH2)2-CNS… M2+ complex. The aforementioned issues are confirmed by donor-acceptor interactions and strongly formed BCPs based on NBO and QTAIM methodologies.
