








1. Background
The human body is the home of more than one trillion microbes with a diverse variety of commensal microbes that play an important role in the health of the individual. These microbes inhabit diverse habitats such as the gut, skin, vagina, oral, etc. The human skin is the largest organ of the human body and plays an important role as the first line of defense against external environmental changes and invading pathogens (1). The skin is an ecosystem composed of microbial communities that inhabit a range of physiologically and topographically distinct niches, including sebaceous/nonsebaceous, hair-bearing/glabrous, moist/dry, and creased/non-creased regions (2, 3). Human microbiome in healthy skin and the overall well-being of the individual has been started to be appreciated since years ago (4). Cataloging the healthy microbiome is a mandatory first step toward identification and correction of the microbial configurations that are implicated in diseases (5). The analysis of the human skin microbiome helps detect the cause behind the occurrence of many complex diseases (6).
2. Objectives
The aim of this study was to identify normal skin microbiome signature of healthy Saudi individuals living in Jeddah, Saudi Arabia through the analysis of 16S rRNA of the resident skin microbiome.
3. Methods
Eight healthy volunteers from Saudi Arabia (4 males and 4 females), aged 20 to 37 years, were enrolled in the current study. The inclusion criteria were no history of dermatologic disorders or current skin infection (atopic dermatitis, psoriasis, and stasis eczema), use of no skin creams or moisturizer before sampling, no treatment with chemotherapy or radiation, or subjects treated with antibiotics within the last three months. Samples were collected by swabbing from inner elbow of the right arm for each subject with no prior cleaning or treatment of skin surface using iSWAB Microbiome Collection kit.
DNAs were extracted using the QIAamp® DNA Microbiome kit (Qiagen®51306; North Rhine-Westphalia, Germany) according to the manufacturer’s instructions. PCR of the V3-V4 regions of bacterial 16S rRNA using 338F and 806R primers was done following standard procedure (e.g., initial denaturation at 95°C for 5 min; 25 cycles of denaturation at 95°C for 30s, annealing at 56°C for 30s, and extension at 72°C for 40s, and final extension of 72°C for 10 min), while deep sequencing was done at Beijing Genome Institute (BGI), China using Illumina platform. Raw sequencing data were deposited in the European Nucleotide Archive (ENA) and received no. PRJNA609106. These data were analyzed using the Quantitative Insights Into Microbial Ecology 2 (QIIME2) package v.2018.11; (https://qiime2.org). Subsequent bioinformatics analysis was done following Abuljadayel et al. (7).
4. Results
4.1. Raw Data Statistics
Statistics of raw data for eight healthy skin microbiome are shown in Table 1, and data were described in Appendices 1 and 2, while results of OTU annotation are shown in Appendix 20. Alpha diversity was applied to analyze the complexity of species. Shannon and Simpson indices (Alpha diversity measures) indicated no significant differences between male and female groups (Appendices 3 and 21). The results in Appendix 4 indicated that F1 and F3 subjects had the lowest richness as referred to by Shannon index, while M4 and F4 showed the highest (Appendix 4). As expected, the data of evenness for Simpson index indicated opposite results (Appendix 4). Plot of principal coordinate analysis (PCoA) (shown in Appendix 5) indicated separation between male and female samples. Rarefaction curves showed that the maximum permitted number of reads for further analysis was ~73,000 (Appendix 6).
| Sample ID | Reads Length (Bp) | Raw Data (Mbp) | N Base (%) | Low Quality (%) | Clean Data (Mbp) | Data Utilization (%) | Raw Reads | Clean Reads | Read Utilization (%) |
|---|---|---|---|---|---|---|---|---|---|
| M1 | 297:297 | 53.65 | 0.049 | 1.966 | 51.51 | 96.02 | 90,315 | 87,239 | 96.59 |
| M2 | 298:297 | 53.71 | 0.047 | 1.713 | 51.74 | 96.34 | 90,266 | 87,448 | 96.88 |
| M3 | 294:297 | 54.52 | 0.051 | 1.823 | 52.46 | 96.22 | 92,256 | 89,228 | 96.72 |
| M4 | 299:297 | 53.89 | 0.071 | 2.011 | 51.62 | 95.78 | 90,425 | 87,122 | 96.35 |
| F1 | 300:297 | 53.27 | 0.087 | 1.627 | 51.38 | 96.45 | 89,235 | 86,529 | 96.97 |
| F2 | 296:297 | 54.33 | 0.041 | 1.911 | 52.26 | 96.19 | 91,616 | 88,602 | 96.71 |
| F3 | 297:300 | 54.50 | 0.067 | 2.373 | 51.62 | 94.72 | 91,285 | 87,485 | 95.84 |
| F4 | 300:297 | 53.68 | 0.076 | 2.044 | 51.46 | 95.87 | 89,911 | 86,656 | 96.38 |
Statistics of Data Generated from Deep Sequencing for Eight Saudi Individuals
4.2. Normal Skin Microbiome Signatures at the Phylum Up to Species Levels.
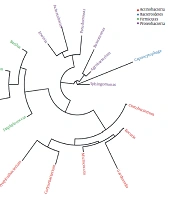
A threshold of ≥ 10 reads was considered highly abundant (Appendix 20) that was met for a number of 21 out of the 28 OTUs (Appendix 22). These OTUs are described in Appendix 23. Phylogenetic tree indicated the existence of four phyla (Figure 1). They include Actinobacteria (six genera), Bacteroidetes (one genus), Firmicutes (four genera), and Proteobacteria (six genera). The results of Appendix 23 align with those of the heat maps at the different taxa levels (Appendices 7 - 12). Venn diagram showed 17 OTUs common in both male and female groups (Appendix 13), while five OTUs were unique in male (Curtobacteriumspp.1, R. mucosa, Corynebacteriumspp.4, Capnocytophaga spp.1, and Mogibacteriaceae, and six in female (Agrobacterium spp.1, Acinetobacter spp.1, Enterococcus spp.1, Gardnerella spp.1, Lactobacillus spp.1 and Corynebacteriumspp.5). The latter results were not considered for further analysis due to the low number of sequences for each OTU (Appendix 13).

Genus level phylogenetic tree of normal skin microbiome. Genera with the same color belong to the same phylum.
4.3. Abundance of Different Microbes Across Sex
Abundance of microbes (weighted Unifrac diversity distances) of different subjects of male and female was studied at the phylum (Appendix 14), class (Appendix 15), order (Appendix 16), family (Appendix 17), genus (Appendix 18) and species levels (Appendix 19). Weighted Unifrac diversity distances showed diversity in different microbiome signatures. Four phyla, four classes, seven orders, 10 families, 10 genera, and two species showed diversity in microbiomes of male and female (Appendices 14 - 19, respectively). The four phyla included Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria (Appendix 14). The four classes included Actinobacteria, Bacilli, and Gammaproteobacteria (Appendix 15).
The seven orders consisted of Actinomycetales, Bacillales, Enterobacteriales, Lactobacillales, Pseudomonadales, Rhizobiales, and Rhodospirillales (Appendix 16). The 10 families included Acetobacteraceae, Bacillaceae, Corynebacteriaceae, Enterobacteriaceae, Enterococcaceae, Microbacteriaceae, Moraxellaceae, Pseudomonadaceae, Rhizobiaceae, and Staphylococcaceae (Appendix 17). The 10 genera included Acinetobacter, Agrobacterium, Bacillus, Corynebacterium, Curtobacterium, Enterococcus, Erwinia, Pseudomonas, Roseomonas, and Staphylococcus. (Appendix 18). The two species included B. cereus and R. mucosa (Appendix 19). Highly abundant OTUs (15 OTUs with ≥ 10 reads) that appeared in all, or in at least three, subjects indicated dramatic difference between sexes. The 15 OTUs referred to either genera or species of which Staphylococcus (spp.1 and spp.2), Erwinia spp.1, Pseudomonas spp.1, Sphingomonas spp.1, Corynebacterium spp.2, Propionibacterium acnes, Kocuria palustris were higher in males (Figure 2), while B. cereus, Bacillus spp.1, Erwinia spp.2, Corynebacterium (spp.1 & spp.3), Micrococcus spp.1, Pseudomonas spp.2 were lower in males (Figure 2).
 and low (B) microbe abundance in male versus female skin microbiome of Saudi individuals. M = male, F = female.")
High (A) and low (B) microbe abundance in male versus female skin microbiome of Saudi individuals. M = male, F = female.
5. Discussion
In the present study, healthy skin microbiome of Saudi residents indicated the presence of as little as six, out of 28 OTUs that were detected at species level. They refer to genera Bacillus (e.g., B. cereus), Roseomonas (e.g., P. mucosa), Kocuria (e.g., K. palustris), Propionibacterium (e.g., P. acnes), Pseudomonas (e.g., P. mendocina), and Corynebacterium (e.g., C. kroppenstedtii), respectively. These six genera belong to phyla Firmicutes (e.g., Bacillus), Proteobacteria (e.g., Roseomonas and Pseudomonas), and Actinobacteria (e.g., Propionibacterium, Kocuria, and Corynebacterium). There is one OTU (e.g., OTU26) that was detected only at family level, e.g., Mogibacteriaceae (Appendix 23). This family is part of phylum Firmicutes.
The other 21 OTUs refer to unassigned species of genera Staphylococcus (Staphylococcus spp.1 and spp.2), Bacillus (Bacillus spp.1), Enterococcus (Enterococcus spp.1), and Lactobacillus (Lactobacillus spp.1) of phylum Firmicutes, Agrobacterium (Agrobacterium spp.1), Sphingomonas (Sphingomonas spp.1), Erwinia (e.g., Erwinia spp.1 and spp.2), Pseudomonas (Pseudomonas spp.1 and spp.2), and Acinetobacter (Acinetobacter spp.1) of phylum Proteobacteria, Curtobacterium (Curtobacterium spp.1), Corynebacterium (Corynebacterium spp.1, spp.2, spp.3, spp.4 and spp.5), Micrococcus (Micrococcus spp.1), Gardnerella (Gardnerella spp.1) of phylum Actinobacteria, and Capnocytophaga (Capnocytophaga spp.1) of phylum Bacteroidetes (Appendix 23).
5.1. Race-specific Healthy Skin Microbiome Signatures
Consistent with Kim et al. (8), healthy skin microbiome in several populations at phylum level are Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidetes. However, our results demonstrated the occurrence of the first three phyla, e.g., Firmicutes (342,875 reads), Actinobacteria (58,322 reads), Proteobacteria (234,757 reads), while low prevalence of Bacteroidetes (2 reads) in healthy skin microbiomes of individuals living in Saudi Arabia (Appendix 23). Results of several other studies (9) align with these of the present study in terms of abundance at the phylum level. Silva et al. (10) also reported an increased level of Firmicutes in healthy skin of different populations. These results align with those of the present study (Appendix 23). Meisel et al. (11) showed that S. epidermidis and S. hominis were prevalent for Staphylococcus. Our results align with those of Kim et al. (8) referring to genus Propionibacterium, while showed no existence of the two Staphylococci species. Interestingly, our results indicated the occurrence of two new unassigned species of Staphylococcus (Appendix 23). The two new species of Staphylococcus might have the same action of the two-missing species in microbiome signature of Saudi individuals.
5.2. Healthy Skin Microbiome and Gender
We suggest that differential abundance of microbes due to gender represents extra environmental factors, influencing such differences in microbiome signature. Previous studies indicated that Propionibacterium, Corynebacterium, and Staphylococcus were more abundant in males (12), while Enterobacteriales, Moraxellaceae, Lactobacillaceae, and Pseudomonadaceae (according to Fierer et al.) (13), and Lactobacillus, Enhydrobacter and Deinococcus (According to Ling et al.) (12) were higher in females. As women use cosmetics more frequently than men (in accordance with Fierer et al.) (13), thereby altering the microbial community structure and diversity of their skin may definitely affect microbe richness and relative abundance compared to men.
In the present study, relative abundances of assigned species of genera Propionibacterium (e.g., Propionibacterium acnes) and Kocuria (e.g., Kocuria palustris) and unassigned species of genera Staphylococcus, Erwinia, Pseudomonas, Sphingomonas, and Corynebacterium are higher in male microbiome, while relative abundances of assigned (e.g., Bacillus cereus), and unassigned species of genus Bacillus and unassigned species of genera Erwinia, Corynebacterium, Micrococcus, Pseudomonas are lower in male microbiome (Appendix 22 and Figure 2). The results of Staphylococcus and Propionibacterium abundances in the present study are in agreement with those of Ling et al. (12) and Fierer et al. (13).
5.3. Conclusions
Overall, the study highlights skin microbiome signature of individuals in Saudi Arabia. This information will be helpful when studying skin microbiomes of patients with atopic dermatitis (AD), Psoriasis, or Acnevulgaris toward the detection of biomarkers of the different diseases.
