


1. Background
Coronaviruses are enveloped single-stranded positive-sense RNA viruses with pleomorphic or spherical forms and projections resembling spikes on the surface, displaying the appearance of a crown (1). The SARS-CoV that was initially found in China in 2002 caused the SARS outbreak in 2002 - 2003. Middle East respiratory syndrome (MERS) was first identified as a serious human disease in Arabian Peninsula in 2012 (2). On December 31, 2019, Wuhan Municipal Health Commission in China discovered a sizable number of pneumonia patients in Wuhan, Hubei (3, 4). This was when the first SARS-CoV-2 outbreak began. The new coronavirus (or SARS-CoV-2) epidemic was classified as a Public Health Emergency of International Concern (PHEIC) by the director general of the World Health Organization (WHO) on January 30, 2020 (5). The WHO reported on January 30 that there were 7818 confirmed SARS-CoV-2 cases worldwide, with 7736 of those cases occurring in China and only 82 cases being related to 18 other countries.
The spike (S) protein of the SARS-CoV-2 is the main protein involved in the interaction of the virus with human angiotensin-converting enzyme2 receptor (ACE2) and transmembrane serine (TMPRSS2) receptors on target cells. Many efforts have been undertaken to create COVID-19 vaccines targeting this viral component (6). Natural selection causes any virus to accumulate random mutations. It has been discovered that the S1 subunit of the "S" protein harbors the majority of alterations responsible for transmission, virulence, and host immune evasion, which besides giving rise to several variations, this phenomenon has numerous therapeutic implications (7, 8).
The most contagious mutant of SARS-CoV-2, delta type (B1.617.2) and delta plus (AY1) strains emerged in the second COVID-19 outbreak wave and were first identified in late 2020 in India, facilitating its rapid spread throughout the globe. The delta plus variant, which was discovered in India on October 5, 2020, was classified later as the variant of concern (VOC) because it was more contagious, could bind lung cell receptors more tightly, and produced a weaker antibody response than delta (9). According to previously published data, a common K-417N mutation between delta plus (AY1) and beta (B1.351) variants was found to be the cause of neutralizing resistance. In Vietnam, another delta variant sub-lineage was detected as delta-V, which caused a rise in virus propagation at the time. As a result of these changes in the "S" protein of the alpha variant, this variant was identified as a hybrid virus.
Omicron (B1.1.529), a novel SARS-CoV-2 strain, was first identified in Gauteng, South Africa, in mid-November 2021 (10). As of December 16, 2021, 87 countries had already discovered the omicron version (11). While the 'S' protein of this mutant strain of SARS-CoV-2 has more than 32 mutations, its receptor binding domain (RBD) revealed 15 alterations (12). Omicron’s rapid rate of transmission, probably due to its ability to hide from the host’s immune system, resulted in a sudden rise in the number of COVID-19 cases worldwide (12). The incubation time of omicron was shorter, lasting just three days (13). The variant caused less damage to the lungs than previously identified strains because it mainly replicated in the upper respiratory tract.
Many scientists believe that omicron’s high transmission rate and very low pathogenicity could lead to the development of herd immunity, providing hope for the end of the pandemic (14). This variant’s moderate pathogenic traits often include "cold and flu" accompanied by a sore throat, headache, myalgia, throat pain, fever, and lethargy, with no signs of a loss of flavor or aroma, as observed in previous SARS-CoV-2 strains. Only a few patients had severe symptoms requiring hospitalization, and most of them experienced mild symptoms that did not demand hospitalization (15).
However, some reports asserted that a significant number (21%) of African patients hospitalized due to contracting the omicron variant had disastrous clinical results (16). The patients infected with the omicron variant developed strong immune responses that could neutralize omicron, as well as other variants of the SARS-CoV-2, reducing the chances of re-infections with delta type, dethroning it as the dominant strain (17). Due to its recent emergence, the omicron variant lacked strong evidence regarding the extent of infection-induced immunity; however, Schulze Zur Wiesch predicted that it would be comparable to other variants. Accordingly, people exposed to omicron in the last few weeks would probably be protected for the upcoming few months given that their antibody levels are high enough because omicron spreads more quickly than earlier strains (18).
2. Objectives
The objectives of this study were to determine the clades, variants, and lineage of SARS-CoV-2 strains extracted from residents and travelers in Duhok, Iraq, by sequencing. Furthermore, we explored the mutational patterns of the viruses circulating in the Duhok population and compared them with those identified in travelers crossing the border from Turkey to trace their epidemiological patterns.
3. Methods
3.1. Study Design and Population
This cross-sectional study was carried out from July 2021 to December 2021. The participants were recruited by random sampling and categorized into two groups. The first group consisted of 700 residents of Duhok city, Iraq, who had not had recent travels. The second group consisted of 700 travelers who crossed the Turkish-Iraqi border of Ibrahim Al-Khalil. All participants had upper or lower respiratory infection symptoms. Exclusion criteria for both groups included previous COVID-19 vaccination and already contracting the natural infection. Nasopharyngeal and oropharyngeal swabs were obtained from all participants and transferred into a viral transport medium (VTM). The samples were kept at -70°C in the Central Public Health Laboratory of Duhok city, Iraq, till further processing.
3.2. Real-time PCR and Viral RNA Extraction
The QIAprep and Viral RNA UM Kit (i.e., Qiagen) was used for the detection and confirmation of the SARS-CoV-2 infection using nasopharyngeal samples. This kit combines RNA extraction with a real-time PCR-based reaction to detect the virus. The procedure was run according to the instructions provided by the manufacturer. Among positive samples for SARS-CoV-2 confirmed by real-time PCR from both groups, selected samples with a cycle threshold < 20 (15 samples from each group) were subjected to RNA extraction utilizing QIAamp Viral RNA Mini Kit from Qiagen. The samples were then shipped on dry ice to the INTERGEN Genetics and Rare Diseases Diagnosis Research and Application Center (Turkey) for next-generation sequencing.
3.3. Bioinformatic Analysis and Next Generation Sequencing
After the samples arrived at Intergen/Turkey, they were re-tested for RNA integrity and target confirmation. For reverse transcription and cDNA synthesis, the Ipsogen RT Kit and nanomere primers (Qiagen, Germany) were employed. Using an Illumina sample preparation kit, individual samples were indexed and tagged, and next-generation sequencing (NGS) was conducted using the Illumina Miseq instrument (Illumina, the US) in accordance with the manufacturer's instructions. Software from the Broad Institute's IGV2.8.9 was utilized to analyze sequencing data, and the BWA-MEM alignment technique was used to assemble and align short-read sequencing with the reference genome (NC045512.2) (19). Furthermore, the annotation of the assembled sequences was conducted using Annovar software according to a previous study (20), followed by data analysis in Lofreq (version 2) for mutation detection and variant calling (21). A quality check was conducted on the sequencing reads that were approved. The sequences analyzed exhibited a coverage rate of > 99% and gaps of 30 bps.
4. Results
Out of 1400 participants, 353 (25.21%) samples from both groups were demonstrated by RT-PCR to be positive. Among thirty samples with Ct values < 20 sent for the SARS-CoV-2/S gene sequencing, we retrieved the complete sequencing of 19 samples, comprising nine samples from Duhok residents and ten samples from travelers. The S gene sequence was obtained and submitted to the GISAID database and allotted the accession numbers of EPI_ISL_13971728 to EPI_ISL_13971746 (Appendix 1).
A total of 76 mutations, including 52 non-synonymous, 16 synonymous, and eight deletional mutations, were detected in the sequenced samples, but no unique mutation was found (Appendix 2). Meanwhile, three well-known international platforms (GISAID, PANGO, and Nextstrain) were widely explored to categorize and separate SARS-CoV-2 isolates into different clades, variations, and lineages (22-24), leading to various naming conventions and the requirement for an all-encompassing unified platform. Using the GISAID system for genetic sequencing to classify isolates, the resulting clades included GK (58%), O (5%), GRA (26%), and G (11%) (Figure 1).

SARS-CoV-2 clades were identified in this study based on sequencing using the GISAID platform.
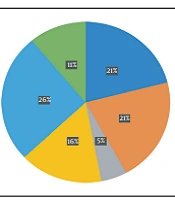
The variants identified were divided into four different groups as follows: 21J (delta), 21K (omicron), 21A (delta), and 20I (alpha V1), each accounting for 53%, 26%, 10%, and 11%, respectively (Figure 2). For sub-classification to lineages and sub-lineages, the PANGO system was utilized, detecting two B.1.1.7, three AY.78, four B1.617.2, four AY.126, one AY.121, and five BA.1.1 variants (Figure 3).

SARS-CoV-2 variants identified in this study based on sequencing using the Nextclade platform.

SARS-CoV-2 lineage identification in this study is based on sequencing using pandemic waves.
4.1. Phylogenetic Analysis
In this study, a total of 48 sequences of the S gene in Iraqi populations and 41 sequences from other countries in the world were retrieved from the GISAID platform. These sequences were used for constructing a phylogenetic tree according to the sequenced isolates that were identified during the second and third waves, as well as at the beginning of the fourth epidemic wave of COVID-19 in the country. The GISAID database was used for phylogenetic evaluations, and a phylogenetic tree was generated (Figures 4 and 5).
. *: Study samples.")
The phylogenetic tree representing the relationship between the SARS-CoV-2 S gene sequences detected in this study and various strains found in Iraqis (SARS-CoV-2 strain grouping was based on GISAID clades). *: Study samples.

The phylogenetic tree represents the relationship between the SARS-CoV-2 S gene sequences identified in this study with the sequences reported from other countries. SARS-CoV-2 strains grouping was based on GISAID clades. *: Study samples.
5. Discussion
With regard to COVID-19, the Iraqi Ministry of Health issued a warning stating that "the epidemiological situation has become dangerous," that the number of infections has been “rapidly increasing,” and that the latest wave, or what it called the “third wave,” was more severe than the previous one. The sharp increase in infections was attributed to peoples’ indifference to preventive measures and lack of compliance with safety regulations, particularly the use of face masks and social distancing. In the present study, we investigated the molecular signatures and patterns of the mutations of the circulating strains of SARS-CoV-2 in Duhok Governorate, Iraq, during the end of the second wave and at the beginning of the third wave of the pandemic, when the number of daily cases reached its highest level since the outbreak began. The worst wave of the pandemic hit the nation at the beginning of the summer of 2021. As a result, a study was necessary to track the evolution of the virus in the country. Compared to other countries, not many studies have been dedicated to tracking and categorizing SARS-CoV-2 variants in Iraq and in the city of Duhok in particular.
The GISAID and Pango lineages used in this study demonstrated that clade G was the most prevalent entity. The dominant genetic amino acid mutation has been found to be D614G, which is found in all sequenced strains. This mutation affects the receptor binding domain of the S spike protein that is responsible for the attachment of the virus to its receptors on host cells. At the same time, this site is where neutralizing antibodies act. In addition, this change seems to enhance the infectivity and stability of SARS-CoV-2 (25).
This study revealed that the GK clade (delta VOC) had the highest prevalence among locals and travelers in Duhok city, followed by the GRA clade (omicron VOC), which was only found in travelers crossing the border from Turkey, confirming the possibility of the transmission of this variant from neighbor countries through traveling. The phylogenetic tree showed high homogeneity between the strains sequenced from Duhok city and the variants registered in GISAID from other countries, including Turkey, Saudi Arabia, Germany, and Switzerland. In contrast to the other variants of SARS-CoV-2, the delta variant first appeared in India in late 2020, which was later found to have greater infectivity and transmissibility (26), as well as a shorter incubation period and the potential to evade neutralizing antibodies (27, 28).
Our analysis of the omicron VOC of SARS-CoV-2 sequences revealed numerous mutations in the S gene, which encodes a structural protein that serves as a viral protein binding to receptors on the host cell’s surfaces and, therefore, determines the host range (29). A total of 31 non-synonymous mutations were discovered in this gene. This gene has been reported to be 4-5 times more likely to become mutated in comparison with other genes of the virus (30). Omicron strains share a dominant polymorphism known as D614-G in the Spike gene, which was discovered in all other VOCs (31). This polymorphism is linked with higher rates of infection and transmission, as well as viral escape from reactive antibodies (32). A mutation hot spot within the spike glycoprotein (known as omicron RBD), which is one of the primary targets for neutralizing antibodies, was shown to include 17 mutations, such as R346K, G339D, S371P, S373P, S371F, S375F, N440K, K417N, S477N, G446S, T478K, Q493R, E484A, Q498R, G496S, Y505H, and N501Y (33).
Based on findings from earlier studies, omicron also possesses numerous novel mutations throughout the gene encoding the spike protein, including in the furin cleavage site, N-terminal domain (NTD), and S2 sub-unit. These mutations may have an impact on the spike protein’s capability to bind the ACE-2 receptor and respond to antibodies (34, 35). Interestingly, the Ins215ped mutation already occurred 127 times (0.00% of all samples with the spike sequence) in 16 countries. The first strain with this amino acid change, identified in December 2021, was hCoV-19/France/ARA-HCL022005965401/2021, and most recently, it was detected in the hCoV-19/England/ALDP-37D7BF2/2022 strain in February 2022.
When compared to other VOCs, the SARS-CoV-2 genome represents a high rate of mutations, particularly those affecting the spike protein, which may boost viral transmission and immunological escape. Additionally, the accumulation of numerous mutations on the immunogenic epitopes of the spike protein necessitates the production of novel vaccines using the omicron as a viable reference strain. More research is necessary to determine the efficacy of current vaccines against the omicron variant. There are a few deletions and more than 30 changes respective to the original sequence in the SARS-CoV-2 omicron variant (36), many of which seem to facilitate the spread of the virus and, actually, are the same mutations found in previous SARS-CoV-2 variants, affecting binding affinity and transmissibility of the virus (37). The consequences of most other omicron mutations are unknown (38). The symptoms caused by the omicron and delta variants are different, and public health and medical professionals need to understand these differences. This not only foretells possible signs to look out for but also helps understand prognosis and outcomes.
5.1. Conclusions
The SARS-CoV-2 outbreak in late 2019 forced all countries’ health systems to pay attention to important public health issues to fight the virus and stop its propagation. The incidence, distribution, and infectivity of COVs have increased in recent years because of the introduction of unique mutations in their genomes. Most novel COVs’ variants have changes in the S1 domain of the spike protein, which is responsible for interaction with ACE2, rendering neutralizing antibodies (monoclonal antibodies, convalescent plasma, and sera from vaccinated people) less effective. Particularly, omicron is an emerging variant with around 30 changes in the amino acid sequence of the spike protein, enhancing its interaction with ACE II and removing the epitopes detected by neutralizing antibodies. Epidemiological monitoring is a crucial tactic for detecting new SARS-CoV-2 variants and characterizing them in terms of the fatality rate. The development of pan-coronavirus vaccines, as alternatives to present vaccines, and monoclonal antibody therapies based on new viral variations can reduce the mortality and morbidity inflicted by the SARS-CoV-2 infection.
