1. Background
Helicobacter pylori, a Gram-negative, spiral-shaped, and microaerophilic bacterium, is a pervasive pathogen infecting approximately half of the world's population. This bacterium is the primary cause of diseases such as stomach ulcers, chronic gastritis, intestinal disorders, and stomach cancer (1-4). Comparative genome sequence analysis of H. pylori strains has revealed significant genetic diversity, primarily due to high mutation rates and genetic recombination in this microorganism (5). This diversity enables H. pylori to adapt to various unfavorable conditions within the host. Furthermore, genotyping studies have highlighted the interplay between microorganisms and host genetics, indicating a potential shared coevolutionary history. The genetic diversity of H. pylori exhibits marked variations across geographical regions and different populations; therefore, it is imperative to conduct regional studies and establish communication with diverse geographical areas (5, 6).
To ascertain the genetic relationship and population structure of H. pylori species, along with evaluating the impact of public health measures on infection control, a fast, inexpensive method with high reproducibility and discrimination power is needed. Several typing methods have been developed to assess the genetic diversity, strain correlations, and epidemiology of H. pylori (7). Among these, Multi locus sequence typing (MLST) and pulsed-field gel electrophoresis (PFGE) are common molecular typing methods. Multi locus sequence typing generates an allelic profile or typing sequence for each isolate, facilitating worldwide sharing through the Internet. However, it is expensive, particularly for characterizing isolates in disease outbreaks (8, 9). In contrast, PFGE, the gold standard in molecular epidemiology, is time-consuming. Multiple-locus variable-number tandem-repeat analysis (MLVA), a PCR-based method, offers comparable strain detection to PFGE with significantly higher discrimination power than MLST. Additionally, MLVA is more cost-effective compared to other methods (10). The MLVA technique relies on identifying the variable number of tandem repeats (VNTR) in specific loci on the microbial genome.
2. Objectives
Considering the aforementioned issues, the present study aims to assess the genetic diversity of H. pylori strains isolated from biopsy specimens using MLVA typing.
3. Methods
The descriptive cross-sectional study was conducted on patients attending Babol University of Medical Sciences, Iran. Seventy non-duplicative biopsy samples were collected from untreated patients undergoing upper gastro-duodenal endoscopy at Ayatollah Rouhani Hospital in Babol, Iran. Ayatollah Rouhani Hospital, affiliated with Babol University of Medical Sciences (BUMS), is one of the most equipped teaching therapeutic centers with over 511 beds. A total of 32 men and 38 women, aged 21 to 89 years (average age: 51.2 ± 1.1 years), presenting clinical manifestations such as abdominal cramps and pain, nausea, vomiting, frequent burping, bloating, and weight loss, were included in the study.
Patients who had received anti-Hp drugs, regimens containing bismuth, proton pump inhibitors (PPIs), non-steroidal anti-inflammatory drugs (NSAIDs), or H2-receptor blockers within four weeks prior to the project or had a history of chronic liver diseases were excluded. Verbal informed consent was obtained from all participants or their companions. All biopsy samples were immediately placed in Stuart’s medium and transferred to a microbiology laboratory within 2 hours, where they were stored at 4°C. After homogenization with an electric tissue homogenizer (Ultraturrax, Iena, Germany), genomic DNA extraction was performed using a kit protocol (Yekta Tajhiz Azma kit, Tehran, Iran).
3.1. Molecular Detection of Helicobacter Pylori
The presence of H. pylori in this strain was confirmed through the PCR method utilizing glmM gene primers (Sina Clone, Iran). The PCR reaction mixture, with a final volume of 25 μL, consisted of 1 μL of DNA extracted from the biopsy sample, 1 μL each of the forward primer (5-GGATAGACGATGTGATAGG-3) and reverse primer (5-TTGGTTAGGGTGTAAAGC-3) (5 pmol/μL each), 2.5 μL buffer (X 10), 1 μL of dNTP (0.2 mM), 0.75 μL of MgCl2 (0.8 mM), 0.75 μL of Taq DNA polymerase (1 unit/μL), and 17 μL of distilled water. The PCR was carried out through 35 cycles using a thermocycler (PCR Thermal Cycler, A&E, China) with the following temperature cycles: Initial denaturation at 95°C for 10 min, followed by denaturation at 94°C for 1 min, annealing at 58°C for 1 min, extension at 72°C for 1 min, and a concluding extension at 72°C for 10 min. Subsequently, the products of the colony PCR reaction were electrophoresed on a 1.5% agarose gel containing ethidium bromide (Cinagen, Iran) at 70 V for 30 min. The gel was then observed under ultraviolet light, and the resulting bands were photographed using a Gel Doc device.
3.2. Helicobacter Pylori Genotyping by Multiple-Locus Variable-Number Tandem-Repeat Analysis Method
In this study, bacterial genotyping was conducted using six VNTR markers (VNTR-180, VNTR-263, VNTR-614, VNTR-607, VNTR-2181, and VNTR-2457), as designed in previous studies by Guo et al. (6) (Table 1). The PCR reaction, with a volume of 25 µL, was prepared by combining 2.5 µL of 10x PCR buffer, 1 µL MgCl2, 0.5 µL dNTP mix, 0.2 µL Taq DNA polymerase enzyme, 1 µL of each primer mentioned above, 2 µL of DNA from each isolate, and 16.8 µL of distilled water. The multiplex PCR reaction followed a thermal and time schedule, including primary denaturation at 95°C for 5 min, followed by 30 cycles. These cycles involved secondary denaturation at 94°C for 45 sec, annealing of primers at 54°C for 40 sec, extension at 72°C for 45 sec, and a concluding extension at 72°C for 7 min. Following the PCR, the product was electrophoresed on a 1% agarose gel. The resulting gel was then photographed and recorded under UV light. After determining the VNTR loci of the H. pylori strains, a phylogenetic tree was generated using PHYLOViZ software. The allelic diversity (AD) of each locus in all studied isolates was calculated using Comparing Partitions online software (http://minisatellites.u-psud.fr/ASPSamp/base_ms/bact).
Table 1.
Characteristics of the 6 Variable Number of Tandem Repeats Loci in the Helicobacter pylori Strains
| Locus Forward and Reverse Primer (F/R) | Length of Fragment | Annealing Temperature (°C) | Product Size Range |
|---|---|---|---|
| VNTR180: F:TAAAGTGAAAGCGTTACAAAAAGAC; R: CTTCAGGGTAGGAATACAGCAGAGT | 180 | 54 | 180 |
| VNTR263: F:TTGAATTGCAAGCTAATGAGTC; R: AGAAGTGTTGATGCTAGAAGAG | 352 | 54 | 352 |
| VNTR614: F:ATTGATTATGATTTTCTTGGCAATTTTG; R: GCTTATGAATGTGTGTTTTGCTGATGAC | 758 | 54 | 370 - 500 |
| VNTR2181: F:TTATGGAAAATATCATACAACCCCCTAT; R: ATTTAGAAAAATTACCCCTTTCATCAAG | 668 | 54 | 378 |
| VNTR2457: F:TAGAAGATTGCTTGAAAAGCCCTTT; R: GCTCTATGATTTTAAAACGCTCCGT | 378 | 58 | 380 - 650 |
| VNTR607: F:GAATTGATTATGATTTTCTTGGCAAT; R: GCTGAAAACGCTAGGGATAGAGC | 650 | 56 | 280 - 500 |
4. Results
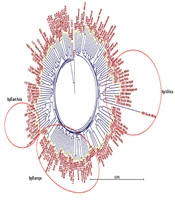
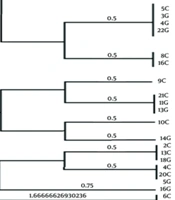
In this study, a total of 70 biopsy specimens were collected from the gastric antrum and gastric corpus of patients referred to Ayatollah Rouhani Hospital in Babol. Out of these, 43 isolates tested positive for the presence of the glmM gene. The PHYLOViZ software was utilized to construct a phylogenetic tree, revealing three clusters and 23 sequence types among all isolates (Figure 1). Upon examining the VNTR diagram for each strain, VNTR-216 and VNTR-218 emerged as the most frequent gene markers across the isolates (Figure 1). The analysis based on Simpson's genetic diversity index identified VNTR-614 as having the highest diversity among all isolates (Table 2) (SDI = 0.512).
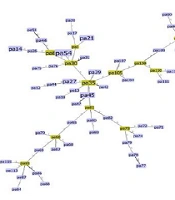
 patterns, as a result, these strains are completely similar. For example, strain 18C is completely similar to strains 19C, 6G, 8G, 10G and 20G, and they are placed in a mini cluster with strain 1C.")
Figure 1.
Evolutionary tree resulting from the analysis of VNTR gene loci of Helicobacter pylori strains. In the phylogenetic tree, some strains have completely similar Multiple-locus variable-number tandem-repeat analysis (MVLA) patterns, as a result, these strains are completely similar. For example, strain 18C is completely similar to strains 19C, 6G, 8G, 10G and 20G, and they are placed in a mini cluster with strain 1C.
Table 2.
Description of 6 Variable Number of Tandem Repeats Loci Analyzing with 70 Helicobacter pylori Clinical Isolates
| Locus | Copy Number | Number of Repeats in VNTRs | Diversity Index |
|---|---|---|---|
| VNTR180 | 6.5 | 20 | 0.450 |
| VNTR263 | 22 | 14 | 0.365 |
| VNTR614 | 6 | 53 | 0.512 |
| VNTR2181 | 26 | 12 | 0.339 |
| VNTR2457 | 10 | 54 | 0.478 |
| VNTR607 | 3 | 138 | 0.498 |
5. Discussion
The prevalence of H. pylori in humans remains high, with more than 50% of the world's population infected (11). A study conducted by Hanafieh and Lopes showed that due to the genetic diversity of H. pylori, the virulence level of this bacterium can be determined by analyzing the population structure and genome sequence of H. pylori strains (12). Notably, a study conducted in western Iran reported a higher prevalence of positive cases at 71%, surpassing previously documented values (13). In Tehran, Bayati et al. found that H. pylori was isolated from biopsy specimens in 32% of participants using the culture method. This variability in infection rates is consistent with findings from studies conducted in different countries (14). The global prevalence of H. pylori infection varies widely, with the highest and lowest rates reported in Nigeria (89.7%) and Yemen (8.9%), respectively. Developing countries exhibit a higher infection rate (50.8%) compared to developed countries (34.7%). Several factors contribute to this disparity, including hygiene, social, and economic conditions. Additionally, the lack of effective methods for purifying drinking water in certain regions, coupled with the reuse of purified water for agricultural, animal husbandry, and industrial purposes in arid and semi-arid areas, may play a role in the transmission of H. pylori to humans.
Certain countries, such as Iran, may provide justifications for the development of diseases at a younger age within their geographical regions (5). However, additional research is essential to ascertain whether changes in the prevalence of H. pylori infection contribute to alterations in the global burden of upper gastrointestinal diseases. In this study, for the first time in Iran, the MLVA technique was used for H. pylori typing. Six loci (VNTR-180, VNTR-263, VNTR-218, VNTR-614, VNTR-245, and VNTR-607) were used to distinguish and determine the type of the strain. This technique was successfully performed and determined the genetic diversity of the strains. This study marks the first implementation of the MLVA technique for H. pylori typing in Iran. Using six loci (VNTR-180, VNTR-263, VNTR-218, VNTR-614, VNTR-245, and VNTR-607), the study successfully distinguished and determined the strain types. The utilization of this technique effectively characterized the genetic diversity among the strains, providing valuable insights into the population dynamics of H. pylori in the region.
The MLVA analysis of 43 isolates from H. pylori gastrointestinal tract infections in patients in Babol revealed the presence of 3 clusters and 23 genotypes. The MLVA-typing method proves effective in distinguishing H. pylori strains, not only among closely related and similar regional strains but also among genetically distant isolates. The results indicated that the AD was lowest at the VNTR-218 locus (0.33%) and highest at the VNTR-614 locus (0.51%). In the study conducted by Guo et al., MLVA was proposed utilizing 12 VNTR loci for H. pylori typing across various regions in China. However, in the current study, we employed six gene loci for genotyping H. pylori strains.
Despite the difference in the number of loci, the results of our study align with those obtained by Guo et al., particularly in terms of genetic diversity (6). In contrast to our study, Sorokin et al. identified 4 clusters and 48 genotypes among 48 H. pylori strains, utilizing the MLVA technique and four VNTR loci. The observed variation in genotypes could be attributed to the diverse geographic origins of strains collected from different regions of Russia, a factor not present in our study where bacterial strains were solely obtained from a single region (7). Additionally, Halicki et al. reported an AD ranging from 0.11 to 0.67 among 90 H. pylori strains from Brazil, using the MLVA technique with four VNTR loci (15).
In this study, the AD of these loci ranged from 0.33 to 0.51, a variation that can be associated with differences in the number of investigated strains. Various studies have consistently shown a high degree of genetic diversity among H. pylori strains in their genome (16, 17). However, the specific relationship between H. pylori genotypes and the associated diseases has not been clearly defined yet. Recently, MLVA, based on changes in the number of repeats at VNTR loci, has been recognized as a valuable molecular tool for genotyping purposes (18, 19). Multiple-locus variable-number tandem-repeat analysis is particularly effective in isolating closely related strains (20). Conversely, the combination of loci with lower variability values proves effective in delineating clear phylogenetic patterns among strains that have evolved over an extended period (21).
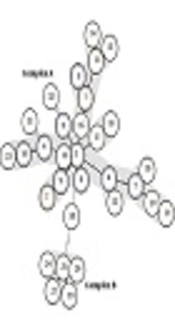
In theory, a higher number of loci results in increased discriminative power, allowing for a more precise establishment of phylogenetic relationships among bacterial strains (6). It is advisable to conduct more extensive studies encompassing diverse geographical populations in Iran to thoroughly explore genetic diversity and elucidate the potential relationship between the geographical origin of strains and the severity of H. pylori pathogenesis. Moreover, H. pylori typing can serve as a valuable tool in delineating human migration patterns and shedding light on the role of ancestral origin in the development of gastric cancer. Among the 6 observed VNTR markers in this study, 5 clonal complexes were identified. Clonal complex CC1, encompassing isolates 18C, 20G, 10G, 6G, 19C, and 8G (with higher allelic similarity), and CC2, including 5C, 4G, 3G, and 22G, revealed that 70% of isolates from these clonal complexes were associated with patients diagnosed with gastritis. This finding suggests an increased likelihood of gastritis in individuals carrying these specific strains. Early diagnosis of patients with these strains is crucial for optimizing disease treatment.
This study has certain limitations: The primary limitation pertains to the incomplete availability of comprehensive patient history background information. Furthermore, it is imperative to acknowledge that the isolation of H. pylori strains was confined to different hospitals, necessitating a cautious approach to the interpretation of the results.
5.1. Conclusions
In conclusion, the elevated AD observed in VNTR 614, 607, and 245 indicates a higher intraspecies diversity of these markers across all isolates in Babol. Consequently, these VNTRs can be deemed ideal loci for typing the studied strains. Overall, the results from this study affirm that the MLVA typing method is successful in achieving molecular epidemiology and geographical classification of H. pylori.