











1. Background
Human papillomavirus (HPV), particularly its high-risk subtypes, has emerged as a critical public health priority due to its well-established etiological role in cervical carcinogenesis. The HPV genotypes 16, 18, 31, 33, 35, 45, 52, and 58 account for approximately 90% of cervical cancer cases globally, representing the most prevalent oncogenic variants (1). Among them, HPV type 52 (HPV52) is the second most common HPV type in cases of high-grade squamous intraepithelial lesions (HSIL) in China (2). Especially in women, the most common high-risk HPV (HR-HPV) type is HPV52 (5.58%) (3). The HPV52 demonstrates oncogenic potential beyond cervical pathology, with emerging evidence linking it to oral carcinogenesis through detectable salivary transmission (4).
Early and accurate identification of HPV subtypes is therefore essential for the effective management and prevention of cervical cancer, as well as for timely therapeutic intervention. Current cervical cancer screening primarily relies on two approaches: Cytologic testing and molecular methods (5). While cytologic testing is limited to patients with visible lesions and fails to identify HPV-infected individuals without lesions, molecular assays face challenges of high cost, limited throughput, and incomplete genotype coverage. These limitations hinder their utility for population-based screening, particularly in resource-limited settings. Consequently, there is an urgent need to develop a cost-effective, high-throughput assay capable of detecting HPV — a critical advancement for improving cervical cancer prevention, especially in developing countries.
HPVs are small dsDNA viruses of approximately 8000 bp in size, with a double-stranded DNA code strand that encodes three regions: The early region (early, E), the late region (late, L), and the non-coding region (NCR) (6). The HPV late region gene encodes the L1 and L2 proteins, which together form the viral capsid, and the HPV L1 protein is extremely conserved and is therefore a reliable basis for identifying different types of HPV (7). We analyzed the crystal structure of HPV52 (PDB ID: 6IGF) and found that the N-terminal 100 Aaa is located mainly on the surface of the protein structure. The L1 protein is an ideal target for the detection of subtype-specific antibodies to HPV, and with a size of approximately 55-60 kDa, it is the main capsid protein of the viral particles (8). The HPV L1 gene has been shown to be the most important protein of the HPV (8).
Previous studies have predominantly employed indirect enzyme-linked immunosorbent assay (ELISA) for HPV antibody detection. However, this method is inherently limited to single antibody subtype detection due to its reliance on enzyme-labeled secondary antibodies, with most research focusing on HPV16 and HPV18 while rarely addressing HPV52 (9-11). In this study, we used a double-antigen sandwich ELISA to detect antibodies to HPV52, which offers better specificity and sample compatibility compared to indirect ELISA methods. The use of L1 partial fragments makes antigen expression and purification easier. This technique offers significant technical advantages in the early identification of HPV52 antibodies and may be useful in the diagnosis and treatment of cervical cancer.
2. Objectives
Given the significance of early and rapid diagnosis of HPV for clinical intervention and treatment, the main objective of this study was to establish a rapid test for infected samples based on the HPV L1 protein truncator for efficient and accurate HPV detection. Secondly, the method is applicable to vaginal douche samples, which is convenient for non-invasive collection and large-scale screening promotion.
3. Methods
3.1. Online Prediction Analysis of Human Papillomavirus L11-141 Epitope Structure
The physicochemical properties of the L11-141 protein were determined using the ProtParam online tool. The linear and conformational B-cell epitopes of L11-141 were predicted using the ElliPro server. The epitope prediction specificity parameter was set to 75% of the system default value, with a threshold set to 0.50 of its default level.
3.2. Cloning of Human Papillomavirus L11-141 Gene Fragment and Construction of pET32a- Human Papillomavirus L11-141 Protein Expression Plasmid
Primers were designed according to the NCBI HPV52 L11-141 gene sequence (JN874436.1). The specific primers for the HPV L11-141 gene were: Forward primer 5’-CGCGGATCCATGGTACAGATTTTAT-3’ and reverse primer 5’-GCGTCGACTTACCTTTTAACCTTTT-3’ (BamH I and Sal I restriction sites are underlined). The prokaryotic expression plasmid, pET-32a HPV L11-141, was constructed and transformed into Escherichia coli DH5α. Positive recombinants were identified through ampicillin-resistant culture and colony PCR, after which plasmids were extracted and subjected to DNA sequencing.
3.3. Expression and Purification of Human Papillomavirus L11-141
The bacteria with the recombinant plasmid were inoculated into 1 mL of LB liquid medium supplemented with ampicillin (100 µg/mL) and incubated overnight at 37°C with shaking (220 rpm) (12). The following day, the entire culture was transferred to 400 mL of fresh LB medium containing ampicillin and grown under the same conditions until the optical density (OD600) reached 0.6 - 0.8. Protein expression was then induced by adding 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG), followed by incubation at 25°C with shaking (100 rpm) overnight. After induction, the cells were harvested by centrifugation at 8000 rpm for 10 minutes at 4°C, resuspended in PBS, and fragmented at 200 W for 4 hours. The inclusion bodies were isolated by centrifugation, solubilized in 8 M urea for 30 minutes, and clarified by additional centrifugation. The supernatant was then subjected to affinity purification using a HisSep Ni-NTA agarose resin, leveraging the His tag encoded by the pET-32a vector for selective binding. Finally, the purified protein was analyzed by 12% SDS-PAGE.
3.4. Horseradish Peroxidase -Labeling of Human Papillomavirus L11-141 Protein
Dissolve 5 mg of horseradish peroxidase (HRP) in 0.5 mL of 0.1 M sodium bicarbonate (NaHCO3), add 0.5 mL of 10 mM sodium periodate (NaIO4), mix, and incubate for 2 hours at room temperature in the dark. Then add 0.75 mL of 0.1 M sodium carbonate (Na2CO3) and 0.75 mL of purified protein, mixing thoroughly. Weigh 0.3 g of Sephadex G25 and place it in a 5 mL syringe with glass wool at the bottom. Add the mixture to the syringe and let it react for 3 hours at room temperature, protected from light. Wash with PBS and collect the eluate. Add 1/20 volume of 5 mg/mL sodium borohydride (NaBH4) solution to the eluate, mix, and react for 30 minutes at room temperature. Add another 3/20 volume of NaBH4 and continue the reaction for an additional hour at room temperature. Finally, wash and collect the cross-links, storing them in small aliquots with equal volumes of glycerol at -20°C.
3.5. Establishment and Optimization of Double Antigen Sandwich Enzyme-Linked Immunosorbent Assay
Various parameters were systematically assessed to optimize working conditions. The most effective concentrations of encapsulated antigen and antibody were determined through checkerboard titration. Different combinations of antigen and enzyme-labeled antigen dilution were tested, and the data was analyzed to identify the most desired response. The exploration of different coating conditions involved incubation at both 37°C for 1 or 2 hours, and at 4°C for 6 or 12 hours, as coating conditions can affect the binding efficiency of antigens or antibodies to the assay plate. Several blocking durations, including 0.5, 1, 2, and 3 hours, were tested to block non-specific binding. The response time for the TMB substrate was optimized at intervals of 5, 10, and 15 minutes. Optimal conditions were obtained by comparing the optical density at 450 nm (OD450) and the positive/negative (P/N) ratio under various experimental conditions and selecting the condition that produced the maximum P/N value.
3.6. Cut-off Value, Diagnostic Sensitivity, and Specificity Determination
Ten negative samples were used to establish the cut-off (CO) value. The method involves measuring the optical density values (OD450nm) at 450 nm for all samples, calculating the mean (X), and setting the CO value to 2X. When the OD450nm measurement reaches or exceeds 2X, the sample is considered positive with a confidence level of 99.9%; conversely, if the OD450nm value is lower than 2X, the result is negative. Female vaginal fluid samples that test positive for HPV are diluted starting at 1:2 and in two-fold increments up to 1:1024. After dilution, the assay was performed using the optimized double antigen sandwich enzyme-linked immunosorbent assay (DAgS-ELISA) method. Under these optimized DAgS-ELISA conditions, the HPV58 positive rate was detected, and HPV52-positive female vaginal wash was used as a positive control to evaluate method specificity.
4. Results
4.1. Epitope Prediction of L1 Protein and Truncated Protein L11-141
The linear epitopes of the L1 protein and truncated protein L11-141 were predicted using BCPred software. The results showed that there were three epitopes above the L11-141 segment, with scores of 1, 0.993, and 0.893, respectively. The epitopes above the L1 protein were found to be multiple and homogeneous. We predicted the epitopes of the L1 protein using Ellipro, and the prediction results indicated that the L1 protein contains abundant B cell epitopes. Therefore, we selected proteins in the 1-141 segment for subsequent experiments (Figure 1).

Epitope prediction of L1 protein and truncated L11-141 and L11-141 protein parameters. A, L1 protein epitope prediction; B, L11-141 protein epitope prediction; C, parameter analysis of L11-141 protein; D, prediction results of L1 protein B cell epitope.
4.2. Identifying Human Papillomavirus-52 L11-141 Gene Amplification Products
The PCR product of the HPV52 L11-141 gene was analyzed by 2% agarose gel electrophoresis and showed a clear fragment of approximately 423 bp, consistent with the predicted size (Figure 2).
 52. Lane M: DNA marker (50 - 500 bp). Lane 1 and 2: HPV type 52 (HPV52) L1<sub>1-141</sub> gene PCR product.")
Electrophoretic profile of PCR product of L1 gene of human papillomavirus (HPV) 52. Lane M: DNA marker (50 - 500 bp). Lane 1 and 2: HPV type 52 (HPV52) L11-141 gene PCR product.
4.3. Expression and Purification of Recombinant Human Papillomavirus-52 L11-141 Fragment
The amino acid sequence encoded by the recombinant expression plasmid pET-32a-HPV52 L1 is identical to that of HPV52 L1 (CZ52A921) recorded in the NCBI database. The induction conditions of HPV52 L1 were explored (Figure 3A). The expression of HPV52 L1 protein in the Rosetta strain of E. coli induced by IPTG, was detected at 34 kDa by SDS-PAGE (Figure 3A). The recombinant HPV52 L1 protein was obtained by sonication of the E. coli Rosetta cells (Figure 3B). The lysed inclusion body of HPV52 L11-141 protein was purified with a Ni-NTA agarose column (Figure 3C and D).
 L1<sub>1-141</sub>. A, SDS-PAGE analysis of expressed HPV52 L1<sub>1-141 </sub>protein. Lane M: Protein marker (8 - 180); lane 1: Uninduced <i>Escherichia coli</i>; lane 2: Induced <i>E. coli</i>; B, western blot analysis of expressed HPV52 L1 protein. His-tag antibody was used as the primary antibody (dilution of 1:2000) ; lane M: Protein marker (8kDa-180); lane 1 and 2: Uninduced <i>E. coli</i>; lane 3 and 4: Supernatant of cell lysate after sonication and centrifugation; lane 5 and 6: Pellet of cell lysate after sonication and centrifugation; C, SDS-PAGE analysis of purified HPV52 L11-141 protein. Lane M: Protein marker (8 - 180); lane 1: Non-purified protein; lane 2: The purified HPV52 L1 protein; D, dot blotting analysis of L1 protein expression in inclusion bodies. Bacterial solution HPV52 L1<sub>1-141</sub> was uninduced (1); bacterial solution HPV52 L1<sub>1-141</sub> induced crushing supernatant (2); bacterial solution HPV52 L1<sub>1-141</sub> induced crushing and precipitation (3).")
Expression and purification of human papillomavirus type 52 (HPV52) L11-141. A, SDS-PAGE analysis of expressed HPV52 L11-141 protein. Lane M: Protein marker (8 - 180); lane 1: Uninduced Escherichia coli; lane 2: Induced E. coli; B, western blot analysis of expressed HPV52 L1 protein. His-tag antibody was used as the primary antibody (dilution of 1:2000) ; lane M: Protein marker (8kDa-180); lane 1 and 2: Uninduced E. coli; lane 3 and 4: Supernatant of cell lysate after sonication and centrifugation; lane 5 and 6: Pellet of cell lysate after sonication and centrifugation; C, SDS-PAGE analysis of purified HPV52 L11-141 protein. Lane M: Protein marker (8 - 180); lane 1: Non-purified protein; lane 2: The purified HPV52 L1 protein; D, dot blotting analysis of L1 protein expression in inclusion bodies. Bacterial solution HPV52 L11-141 was uninduced (1); bacterial solution HPV52 L11-141 induced crushing supernatant (2); bacterial solution HPV52 L11-141 induced crushing and precipitation (3).
4.4. Optimization of Double Antigen Sandwich Enzyme-Linked Immunosorbent Assay Conditions
The optimal reaction conditions for the DAgS-ELISA method were determined through controlled-variable experiments: Firstly, the antigen encapsulation concentration was precisely set at 20 μg/mL (Figure 4); and secondly, the dilution ratio of the enzyme-labeled antigen was set at 1:200 (Figure 4).
 conditions.")
Optimization of double antigen sandwich enzyme-linked immunosorbent assay (DAgS-ELISA) conditions.
4.5. Cut-off Value of Double Antigen Sandwich Enzyme-Linked Immunosorbent Assay
To establish the CO value for a positive outcome, 10 samples of HPV-negative female vaginal wash were processed using the optimized DAgS-ELISA technique. The CO value is determined as the mean OD450nm value of the samples multiplied by 2. For female vaginal wash samples, if the OD450nm value reaches or exceeds 0.072, the sample is considered positive; otherwise, it is considered negative.
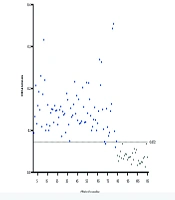
4.6. Detection Effect of Double Antigen Sandwich Enzyme-Linked Immunosorbent Assay
The established DAgS-ELISA method was used to detect 115 clinical female vaginal wash specimens from Yunnan First People's Hospital (Figure 5), with 84 of these samples testing positive and 31 testing negative. Each well plate was coated with 20 μg/mL protein, and the detection antigen labeled with HRP was diluted at 1:500. The actual number of positive samples detected was 79, with all negative samples correctly identified. This resulted in a calculated sensitivity of 94% and a specificity of 100% for this method (Table 1).

Clinical sample test results.
| Methods | PCR Test | Total | |
|---|---|---|---|
| Postive | Negative | ||
| DAgS-ELISA | |||
| Positive | 79 | 0 | 79 |
| Negative | 5 | 31 | 36 |
| Total | 84 | 31 | - |
Abbreviation: DAgS-ELISA, double antigen sandwich enzyme-linked immunosorbent assay.
5. Discussion
Human papillomavirus, as the most common sexually transmitted infection worldwide, is a key etiological factor in the development of cervical cancer when persistent infection occurs (12, 13). Although most HPV infections are self-limiting, persistent infection with HR-HPV can lead to intraepithelial neoplasia and progression to malignancy (14). According to the WHO, low- and middle-income countries bear more than 90% of the global burden of cervical cancer due to a lack of effective screening and treatment (15). In China, HPV type 52, the fourth most predominantly infected subtype, is detected in cervical cancer patients at a significantly higher rate than in other regions, highlighting an urgent need for targeted prevention and control strategies (16, 17).
Currently, HPV antibody detection techniques mainly include neutralization tests and ELISA (18). Although the neutralization test can effectively assess the level of humoral immune response, it is difficult to meet the demand for large-scale clinical screening due to its complicated operation procedure and long detection period (19). The double antigen sandwich ELISA technique provides a new direction for HPV antibody detection by virtue of its high specific antibody recognition ability and the advantage of early serological conversion detection (20). However, this technique has not yet been applied to HPV type 52 antibody detection, and the correlation between clinical sensitivity and pathological changes in the disease still needs to be verified (21).
In addition, there is a mismatch between the sensitivity threshold of current HPV detection technology and clinical needs, such as the mismatch between the clinical sensitivity of 5000 copies/mL in the HC2 assay system and the ultra-high analytical sensitivity in the PCR assay, suggesting an urgent need for the development of detection solutions that are more relevant to clinical diagnostic needs (22, 23). Based on this, this study aims to overcome the technical bottleneck of HPV52 antibody detection, focusing on the development of an HPV52-specific antibody assay based on the principle of double-antigen sandwich ELISA. This assay aims to achieve high sensitivity, high specificity, and economic applicability through the optimization of antigen design and the reaction system to satisfy the needs of primary healthcare and large-scale screening.
Secondly, a clinical sample validation study was conducted to systematically evaluate the antibody recognition ability of the test in patients infected with different HPV52 genotypes and to verify the reliability of the test. This study also has several shortcomings, such as the insufficient sample size of the test, and the collection of samples may result in large sample variation due to manipulation. Therefore, the method should be combined with histopathological diagnosis to improve its clinical value in cervical precancerous lesion screening and provide accurate and efficient technical support for the early prevention and control of cervical cancer.
Additionally, in the selection of epitopes of the L1 protein antigen, although the selected region is close to the surface of the protein, which is favorable for antibody detection, the antigenicity of this region is not the strongest, which is a limitation of this study. In the future, further studies could consider other highly antigenic regions of the L1 protein, and the type and number of clinical samples could be further expanded for the detection method established in this study, to achieve early use for clinical testing.
5.1. Conclusions
Taken together, this study provides a high-throughput, rapid assay for antibody surveillance of HPV infection and immunization, which can serve as a useful complement to traditional screening methods.
