




1. Background
Most people in the world have a history of infection with Helicobacter pylori, a Gram-negative, spiral-shaped, and microaerophilic bacillus that resides the human gastric and duodenal mucosa. Persistent, chronic infection with H. pylori induces stomach ulcers and gastric cancer (1-4). Helicobacter pylori is recognized as a class I human carcinogenic agent and one of the etiological agents in human gastric adenocarcinoma (5). Thus, it seems that prevention, early detection, and treatment of this infection can decrease morbidity and mortality rates in infected patients. Unfortunately, current treatment against H. pylori infection is associated with some drawbacks such as re-infection, increased antibiotic resistance, side effects, patient compliance, and high cost (6, 7). Therefore, vaccination against this bacterium could be a potential way to control the H. pylori infection.
Epitope-based vaccines are a new strategy for increasing immune response to pathogens. Some advantages of an epitope vaccine include (i) increased safety, (ii) increased immunogenicity of predefined epitopes, and (iii) ability to focus immune responses on conserved epitopes (8). However, research has proven that immunity created against H. pylori using a single antigen does not lead to effective protection, rather effective immunity against this infection is achieved via a combination of various antigens (9-11). Some commercial diagnostic kits used to detect H. pylori infection utilize a combination of antigens because this strategy provides high sensitivity and specificity compared to a single antigen (12-15).
Based on previous studies, UreB and FlaA are excellent candidates for the production of vaccines and diagnostic antigens. UreB (the B subunit of urease and one of the four subunits of this enzyme) elicits an immune response; it acts as a portion of whole urease to metabolize urea to ammonia and generate a neutral environment (16-20). FlaA (the predominant subunit of the flagella) is an important pathogenic factor responsible for H. pylori colonization and persistent infection (7, 21-23).
2. Objectives
In this study, we designed, constructed, cloned, and expressed a recombinant multi-epitope gene based on the combined immunodominant regions of UreB and FlaA (rFlaA-UreB) genes of H. pylori. Moreover, we detected its antigenicity as a vaccine candidate and diagnostic antigen by using infected human sera in western blot analysis.
3. Methods
3.1. Chromosomal DNA of H. pylori, Strains, Plasmids, and Media
Chromosomal DNA of H. pylori was purchased from Imam Khomeini Hospital (a teaching university hospital in Tehran, Iran). Escherichia coli strain DH5α (Stratagene, USA) was used for cloning. The recombinant protein was produced by pET32a plasmid (Novagene, USA). The FlaA-UreB recombinant protein was produced using pET32a in E. coli BL21 (DE3) pLysS.
3.2. Identification of Antigenic Regions
We used UreB (NC-018939.1) and FlaA (WP-000885496.1) gene sequences for epitope mapping. B-cell epitope prediction was performed using IEDB, ElliPro, DiscoTope, and IgPred (24).
3.3. Gene Replication
Specific primers required for polymerase chain reaction (PCR) were designed using Allele ID software. The FlaA gene of H. pylori was amplified by primers 5’CGGGATCCGGCGTGTTAGCAGAAGTGAT3’ (forward primer) and 5’CGCCGCCGCCAGGACATTGAGCTCTTAGCGTC3’ (reverse primer) with a linker sequence. The UreB gene was amplified by 5’CTGGCGGCGGCGGCTATGGGTCGTGTGGGT3’ (forward primer) and 5’CCGCTCGAGGAAAATGCTAAAGAGTTGCG3’ (reverse primer) with a linker sequence. The replication of the FlaA gene was done in a 25-µL volume, containing 1 µL of template, 10 pmol of each primer, 50 mM of MgCl2, 0.5 µL of dNTP, 2.5 µL of buffer (10×), and 1 unit of Taq polymerase. Proliferation of the FlaA gene was done under the following conditions: initial denaturation at 95°C for 5 minutes, followed by 30 cycles of denaturation at 95°C for 20 seconds, annealing at 56°C for 20 seconds, extension at 72°C for 40 seconds, and a final extension at 72°C for 5 minutes.
The PCR of the UreB gene was performed in a total volume of 25 µL under the same PCR conditions as the FlaA PCR amplification (except for annealing at 62°C for 20 seconds) (25). Then, for making the conjugated gene as a template for last PCR amplification, we prepared a tube in a total volume of 25 µL, containing 1.5 µL of FlaA PCR product, 1.5 µL of UreB PCR product, 1.5 µL of MgCl2 (50 mM), 0.5 µL of dNTP, 2.5 µL of buffer (10×), and 0.5 µL of the Taq polymerase (neither of primers was used) and amplified by 20 cycles in the same PCR conditions as the FlaA and UreB PCR amplification (except for annealing at 55°C for 20 seconds). The final PCR was done in a 25-µL volume containing 2 µL of template (1 to 6 diluted previous PCR product in double distilled water), 0.5 µL of forward primer of FlaA, 0.5 µL of reverse primer of UreB (10 pmol), 1.5 µL of MgCl2 (50 mM), 0.5 µL of dNTP, 2.5 µL of buffer (10×), and 0.5 µL of the Taq polymerase. The final duplication was performed at an annealing temperature of 58°C.
3.4. Recombinant Plasmid Construction and Purification
The PCR product, rFlaA-UreB, was purified by the high-pure PCR product purification kit (Roche, Germany). Both pET32a and PCR product were digested with BamHI and XhoI and ligated by T4 DNA ligase (Cinagene, Iran). Moreover, E. coli BL21 (DE3) pLysS (Stratagene, USA) and E. coli DH5α (Stratagene, USA) component cells were prepared using calcium chloride (CaCl2) method (25) and transformed by plasmids. The pET32a-FlaA-UreB was transformed into E. coli BL21 (DE3) pLysS cells as the host for the expression of recombinant protein production (25). To confirm the transformation of pET32a-FlaA-UreB into E. coli BL21 (DE3) pLysS, the PCR and enzymatic digestion with BamHI and XhoI were performed. Finally, the nucleotide sequences of recombinant plasmid pET32a-FlaA-UreB were analyzed using the dideoxy chain termination method by standard primers, according to a procedure described by Sanger et al. (26).
3.5. Expression of Recombinant Dual Antigen Multi-Epitope Protein rFlaA-UreB
The pET32a-FlaA-UreB plasmids were transformed into E. coli BL21 (DE3) pLysS grown in 50 mL of nutrient broth supplemented with ampicillin (100 µ/mL) and chloramphenicol (35 µg/mL) at 37°C with vigorous agitation at 220 rpm. The cultured cells were grown until OD600 nm of 0.6. The production of rFlaA-UreB recombinant protein was induced by adding 50 µg of Isopropyl-β-D thiogalactopyranoside (IPTG) to the final concentration of 1 mM, followed by 4 hours of incubation. The recombinant protein was purified with Ni-NTA Agarose resin (Qiagen, USA) based on the manufacturer’s instruction. The purified recombinant protein was analyzed by SDS-PAGE (15%) and spectrophotometry (260/280 nm) (25).
3.6. Immunoblot Analysis
Immunoblot analysis was done (25) using the sera obtained from 15 seropositive H. pylori patients and 15 negative human sera detected by culture, urease test, and pathological analysis. For immunoblot analysis, 0.5 µg of the purified recombinant protein was used per well. Then, the recombinant protein was blotted on polyvinylidene difluoride (PVDF) membrane (Roche, Germany) using transfer buffer containing 25 mM of Tris (pH = 8.3), 192 mM of glycine, and 20% methanol at 90 V for 3 hours at 4°C. The blotted PVDF was blocked with 2.5% (w/v) BSA in TBS buffer for one hour at room temperature. Then, PVDF was incubated for 2 hours at room temperature with diluted (1:100) and normal patients sera. After reaction with the primary antibody, the blot was washed three times with Tris-buffered saline (TBS; 0.5 M of NaCl, 0.02 M of Tris, and 0.05% Tween-20, pH = 8.5) and incubated with anti-human IgG (Abcam, United Kingdom) in 1:1000 dilution. The blots were developed by diaminobenzidine (DAB) solution (Sigma, USA).
3.7. Statistical Analysis
Statistical analysis was performed using t test to evaluate differences in variables between the groups. Comparison between groups was made based on the Mann-Whitney U test. Data were analyzed by SPSS version 16.0 software.
4. Results
4.1. Detection of Antigenic Region
In this study, we predicted antigenic fragments of FlaA and UreB protein sequences through epitope mapping. We used IEDB software to predict the B-cell epitope. According to the results, amino acid sequences of 215 to 352 in FlaA (Figure 1) and 365 to 568 in UreB (Figure 2) were selected as regions with the most pronounced antigenic properties.

The result of FlaA antigenic fragment prediction

The result of UreB antigenic fragment prediction
4.2. DNA Amplification, Multi-Epitope Gene Construction, and Molecular Characterization of rFlaA-UreB
The amplified genes had the expected size of 414 base pair (bp) in FlaA and 609 bp in UreB compared to a 100-bp DNA ladder (Figure 3). The rFlaA-UreB gene was constructed with 1035 bp compared to a 100-bp DNA ladder (Figure 4). The results of PCR and enzymatic digestion of the recombinant plasmid (pET32a-FlaA-UreB) with restriction enzymes confirmed the transformation and showed that the target gene was inserted into the vector correctly. Then, pET32a-FlaA-UreB was sequenced and the results were confirmed. Data showed that the target gene had 100% homology with the reported original sequences (data not shown).

FlaA and UreB genes from Helicobacter pylori amplified by PCR. Lane 1: negative control of UreB, lane 2: positive control of UreB, lane 3: UreB gene, lane 4: 100-bp DNA marker, lane 5: FlaA gene, lane 6: negative control.

rFlaA-UreB recombinant multi-epitope gene construction and amplification by PCR. Lane 1: negative control of rFlaA-UreB, lane 2: 100-bp DNA marker, lane 3: rFlaA-UreB PCR product.
4.3. Expression and Purification of Recombinant Protein
The molecular weight of the recombinant protein was estimated at 38.045 kDa. In the presence of 6xHis-tag in the N-terminal region of rFlaA-UreB added by the pET-32a vector, the protein molecular weight, purified using Ni-NTA resin affinity chromatography, was 57.62 kDa. The SDS-PAGE analysis of induced E. coli BL21 (DE3) pLysS showed a band similar to what that had been predicted with desired molecular weight (Figure 5).
 pLysS</i> destruction pellet, lane 2: <i>pET-32a-FlaA-UreB</i> after 2-h induction, lane 3: protein marker, lane 4 - 7: purified <i>rFlaA-UreB</i> protein.")
Expression and purification of rFlaA-UreB protein. Lane 1: Escherichia coli BL21 (DE3) pLysS destruction pellet, lane 2: pET-32a-FlaA-UreB after 2-h induction, lane 3: protein marker, lane 4 - 7: purified rFlaA-UreB protein.
4.4. Immunoblotting Analysis
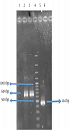
In this study, the antigenicity of the recombinant protein was determined by Western blotting via the sera of humans who suffered H. pylori infection. Our data showed the desired band with an estimated molecular weight of 57.62 kDa. In fact, the recombinant protein reacted with the patients’ sera whereas all 15 human normal sera used as negative controls failed to act similarly. Therefore, based on Western blot analysis results, the produced recombinant protein had an antigenicity enabling it to react with the sera of patients (Figure 6).
.")
Western Blot analysis of rFlaA-UreB by using infected human sera. Lanes 1 - 4: Western Blotting by infected human sera, lane 5: protein marker, lane 6: Western Blotting by normal sera (negative control).
5. Discussion
Nowadays, the technology of multi-epitope antigens is widespread. Design and construction of multi-epitope DNA and protein antigens can serve as (i) a vaccine candidate against different cancers like breast cancer and cervical cancer (27, 28), (ii) a vaccine candidate for the prevention and control of some infectious diseases like Toxoplasmosis, Influenza, and HIV-1 (29-31), and (iii) an antigen in diagnostic kits for Trypanosoma cruzi, Toxoplasma gondii, Hepatitis C, and Tuberculosis (32-35). Several virulence genes of H. pylori have been identified, some of which including UreB, HpaA, NapA, FlaA, and FlaB have been investigated as vaccines and serologic diagnostic candidates for H. pylori infection (36-39).
The prevalence of these antigens in H. pylori isolated strains is 100%, 100%, 93.6%, 100%, and 99%, respectively. Therefore, among proteins, UreB is the optimal antigen and others can be potential antigens for developing H. pylori diagnostic kits and vaccines (40). The benefits of this protein in stimulating immune responses include the same nucleotide sequence and high prevalence among strains, high molecular weight, high expression rate, and strong antigenicity of urea protein (41). Previous research on this antigen showed despite these benefits, oral immunization with a dual antigen was achieved using a combination of H. pylori heat-shock protein and urease in H. felis-infected mice models and induced a strong immune response.
Similarly, in another study, immunization with HpaA or UreB alone induced weak immune responses; however, when both antigens were used together, a stronger immune response was produced (42-45). These results indicate that combining antigens can be a good strategy for developing vaccines against H. pylori infection. Moreover, a mixture of antigens in diagnostic kits provides higher sensitivity and specificity than a single antigen (12-15). The 100% FlaA expression rate among different H. pylori isolates and 98.4% of its specific antibody positive rate in infected patients (with the highest expression rate and specific antibody positive rate in the second rank after UreB) make it appropriate for selection compared to other antigens of H. pylori (40).
The construction of many epitope proteins involves several proteins or domains of proteins. The linker sequence is particularly important for the construction of functional fusion proteins. Several studies have investigated the linker sequence, showing that the flexibility and hydrophilicity of the linker were important in the function of domains. For this reason, we chose LAAA as a peptide linker for making a recombinant multi-epitope protein (46).
Therefore, in this study, we decided to design and construct a multi-epitope antigen via combining the hyperantigenic regions of UreB with the hyperantigenic regions of FlaA in order to increase the specific immune response and safety and decrease side effects of unfavorable epitopes associated with complete antigens. Conserved epitopes of UreB and FlaA with appropriate antigen properties were determined by bioinformatics methods and used instead of whole antigens (11). These regions were confirmed by four software applications, as mentioned earlier. Based on the results, the recombinant protein had high antigenicity and could stimulate the immune response.
In this study, the antigenicity of the specified regions was evaluated through the production of recombinant rFlaA-UreB. Then, the antigenicity of the protein was examined by immunoblotting, performed on the patients’ sera with H. pylori infection. Therefore, it can be concluded although rFlaA-UreB is much smaller than normal UreB and FlaA, it has the same antigenic properties. Thus, it seems this recombinant protein can be effectively used for vaccines. Moreover, it could be applied as a diagnostic antigen for H. pylori infections in different kits.
5.1. Conclusions
In summary, the study identified the antigenic areas of recombinant rFlaA-UreB protein by human sera infected with H. pylori. It can be concluded that the antigenic areas of this multi-epitope protein have an antigenic property that can be further used for the development of H. pylori vaccines and diagnostic kits.
