






1. Background
2. Objectives
3. Methods
3.1. Specimen Preparation and Viral DNA Extraction
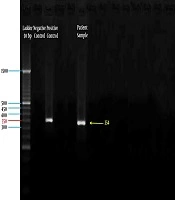
3.2. Amplification of HBoV Genomic DNA
| Primer | Sequence (5’→3’) | Locationa | Size, bp |
|---|---|---|---|
| GSP1 | GCG TTC AAC GCG TCT GAG TAG | 757 - 777 | |
| GSP2 | GCT CCT CCA ACA AGT ATG TGA | 603 - 623 | |
| Nested GSP | CTA GTG AGC ATA AAA TAT CT | 444 - 464 | |
| ORF1-1F | GCG TGG TGA GTG ACA CTA TGG CC | 239 - 262 | 612 |
| ORF1-1R | CAT GAA GGT CAC CTC GCT TGT CTC | 826 - 850 | |
| ORF1-2F | CAT GGT CAG GGC ACA CTG GTA AC | 782 - 805 | 797 |
| ORF1-2R | RCG AAC GCC TTG RAC TAT GG | 1558 - 1578 | |
| ORF1-3F | CGC CAT CTG CTG TGT ACT TAA C | 1464 - 1486 | 830 |
| ORF1-3R | GAT TAT CCA CGT TCG ATG CCT CC | 2270 - 2293 | |
| ORF1/ORF2-F | CAT TCA CAG GAC TAC ACG CTT C | 2038 - 2060 | 376 |
| ORF1/ORF2-R | GGA GCT CAT CTT CGT CTC TAG G | 2391 - 2413 | |
| ORF2-F | CAA CCT AGA GAC GAA GAT GAG C | 2389 - 2411 | 617 |
| ORF2-R | CTT CGT CTG TTA CCT CCT CTG | 2984 - 3005 | |
| ORF3-1F | CAG GAA TCA GAG GAG GTA ACA G | 2978 - 3000 | 737 |
| ORF3-1R | AAT ATG ACC AHG GTG TGC TKA CG | 3691 - 3714 | |
| ORF3-2F | GAG GCA GCT AYT TYA CTG AYT C | 3559 - 3581 | 704 |
| ORF3-2R | TGA TGC TGT GYT TCC GTG YTG TC | 4239 - 4262 | |
| ORF3-3F | GAY RTV ATG CCA GAA CTT CC | 3948 - 3968 | 634 |
| ORF3-3R | TTC CTC TGT AGA GAG CTT GRT C | 4559 - 4581 | |
| ORF3-4F | CCA YCA YTR TCC ATG CTY AGA GAC | 4539 - 4563 | 615 |
| ORF3-4R | CAT CGG ACT RTA GCC TCG AAC TY | 5156 - 5153 | |
| 3′-Oligo(dT)-anchor-R | GTT CCT CTC CAA TGG ACA AGA GGA TTT TTT TTT TTT TT | 3′-end poly(A) tail | |
| 3′-Anchor-R | GTT CCT CTC CAA TGG ACA AGA GGA |
Abbreviation: GSP, gene-specific primer; ORF, open-reading frame.
aAccording to GenBank accession number GU048663.
3.3. Analysis of the 5’ and 3’-ends of the HBoV Genome
3.4. Cloning and Sequencing of the Complete Genome
3.5. Phylogenetic Analysis
| Strain | Accession No. | Genotype | Nucleotide Sequence Similarity, % | Amino Acid Similarity, % | |||||
|---|---|---|---|---|---|---|---|---|---|
| Full-Length | NS1 | NP1 | VP1/VP2 | NS1 | NP1 | VP1/VP2 | |||
| CBJ030 | KM464730 | HBoV1 | 76.3 | 74.2 | 76.4 | 79.0 | 77.6 | 69.8 | 79.9 |
| HZ1402 | KP710212 | HBoV1 | 76.3 | 74.3 | 76.2 | 78.9 | 77.6 | 69.8 | 80.0 |
| KU3 | JQ411251 | HBoV1 | 76.3 | 74.4 | 76.4 | 78.7 | 77.6 | 69.8 | 79.7 |
| Mty1117 | KX373885 | HBoV1 | 76.3 | 74.4 | 76.4 | 78.8 | 77.6 | 69.8 | 79.7 |
| P214 | KX373884 | HBoV1 | 76.2 | 74.4 | 76.4 | 78.5 | 77.6 | 69.8 | 79.9 |
| Nsc10-N386 | JQ964116 | HBoV2A | 99.2 | 99.0 | 99.4 | 99.5 | 99.5 | 100 | 99.9 |
| UK-648 | FJ170280 | HBoV2A | 98.9 | 98.7 | 99.4 | 99.0 | 99.5 | 100 | 99.4 |
| W153 | EU082213 | HBoV2A | 98.9 | 98.8 | 99.4 | 99.0 | 99.5 | 100 | 99.6 |
| CU54TH | GU048663 | HBoV2B | 97.5 | 98.1 | 99.7 | 96.2 | 99.2 | 100 | 97.5 |
| LZFB080 | KM624025 | HBoV2B | 97.0 | 97.5 | 99.5 | 95.6 | 99.1 | 100 | 97.2 |
| NI-327 | FJ973559 | HBoV2B | 96.0 | 97.6 | 96.6 | 94.2 | 99.2 | 99.1 | 96.4 |
| NI-213 | FJ973560 | HBoV2B | 95.8 | 97.4 | 96.8 | 94.2 | 99.2 | 99.1 | 96.3 |
| SH3 | FJ375129 | HBoV2B | 96.6 | 97.2 | 97.5 | 95.6 | 98.9 | 98.6 | 98.8 |
| 46-BJ07 | HM132056 | HBoV3 | 79.9 | 74.5 | 75.8 | 87.7 | 74.3 | 70.2 | 90.9 |
| CU2139UK | GU048665 | HBoV3 | 80.0 | 74.5 | 76.2 | 87.6 | 74.3 | 70.2 | 90.6 |
| TUA21007 | FJ973562 | HBoV3 | 80.0 | 74.5 | 76.1 | 87.7 | 74.3 | 69.8 | 90.9 |
| W471 | NC012564 | HBoV3 | 80.0 | 74.5 | 76.2 | 87.7 | 74.3 | 70.2 | 90.7 |
| W855 | FJ948861 | HBoV3 | 80.0 | 74.6 | 76.1 | 87.6 | 74.3 | 69.8 | 90.7 |
| CMHS01111 | KC461233 | HBoV4 | 89.4 | 91.4 | 92.0 | 87.1 | 93.0 | 89.8 | 90.4 |
| HBoV4-NI-385 | FJ973561 | HBoV4 | 88.6 | 90.4 | 87.8 | 87.6 | 90.6 | 83.3 | 90.3 |
Abbreviation: HBoV, Human bocavirus; NS, non-structural; VP, viralcapsid proteins.
4. Results
4.1. Phylogenetic and Similarity Analysis
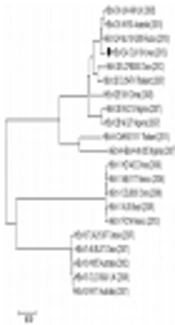
”. The closed circle indicates the novel HBoV CUK18 strain analyzed in this study.")
Phylogenetic analysis of the complete genome sequences of HBoV CUK18 and reference strains. Representative strains are referred to by “genotype strain country (detection year)”. The closed circle indicates the novel HBoV CUK18 strain analyzed in this study.
 and amino acid (B) sequences of NS1, NP1, and VP1 of HBoV CUK18 and reference strains. Representative strains are referred to by “genotype strain”. Closed circles indicate the novel HBoV CUK18 strain analyzed in this study.")
Phylogenetic analysis of the nucleotide (A) and amino acid (B) sequences of NS1, NP1, and VP1 of HBoV CUK18 and reference strains. Representative strains are referred to by “genotype strain”. Closed circles indicate the novel HBoV CUK18 strain analyzed in this study.
4.2. Open Reading Frame Analysis

Comparison amino acid substitutions in ORF3 among HBoV genotypes. Two representative strains were selected for each HBoV genotype. Red vertical arrows indicate sites of amino acid substitutions. Amino acid sequences that differ among viral strains are highlighted.