





1. Background
Hepatitis C virus (HCV) belongs to the genus Hepacivirus and family Flaviviridae. Genetic material of HCV contains an Open Reading Frame (ORF) of almost 9400 nucleotides flanked by Untranslated Regions (UTR) at 5’ and 3’ (1). Hepatitis C virus is a single-stranded, positive polarity RNA. The translation is initiated by IRES at the 5’UTR in endoplasmic reticulum. A single polyprotein precursor is processed into ten proteins by cellular and viral proteases. The essential components are the structural proteins (core, E1, E2), and p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B are seven nonstructural proteins (2). Based on nucleotide heterogeneity, HCV is classified into different genotypes. In Pakistan, genotype 3a is the predominant genotype (3). High genetic heterogeneity characteristic of HCV is the result of error-prone RNA dependent RNA polymerase (RdRP), which is a significant problem for future vaccine development (4). These genotypes are further divided into closely akin multiple sub-types. These subtypes are different from each other by 15% in nucleotide sequences (5, 6).
A large number of closely related viral variants are continuously produced in this progressive disease due to mutation at the nucleotide level and higher multiplication rate of HCV. In vivo, these variants circulate as quasispecies (5, 7). Geographically, genotypes of HCV have distinctive distributions. It seems HCV genotypes 1, 2, and 3 are present globally, but their relative presence varies (8). Different genotypes have different clinical management and epidemiology. The cardinal way of identification and classification of new HCV genotypes can be measured by pairwise similarities in sequence or by phylogenetic study of nucleotide sequences (9). Core protein can form topological identical trees to those determined from the analysis of complete genomic sequences (10). Despite the limited power for phylogenetic analysis, previous studies have selected core gene for determining evolutionary relationships among viral isolates (11, 12). The core protein of HCV is an RNA binding basic protein, which forms a viral capsid. It is released as a 191 amino acid (aa) precursor of around 23-kDa molecular weight containing three conserved domains (122 aa N terminal hydrophilic domain, 50 aa hydrophobic C terminal domain, and 20 aa signal peptide domain).
Besides viral capsid formation, it also interacts with various cellular proteins and pathways that play an essential role in the HCV life cycle (13). Many studies have demonstrated that the highly conserved HCV core protein plays an important role in the pathogenesis and progression of the disease due to its ability to interact with a wide spectrum of viral and cellular proteins, including protooncogenes. Particularly, amino acid substitutions at positions 70 and 91 in the HCV core region have been identified as predictors of hepatocellular carcinoma (HCC) among genotype 1b-infected patients from Japan (14). Hence, these polymorphisms might be considered surrogate markers for hepatic disease stages or the eventual development to HCC (15). Although there is consensus on HCV genotype 3a prevalence in Pakistan, variable frequencies of other HCV genotypes have been reported (16). A phylogenetic study has a central role in understanding evolution, ecology, biodiversity, and viral genomics.
Identification and naming of organisms are performed by phylogenetic analysis. Distribution and genetic diversity of the HCV genome are poorly documented in non-western countries. Efficient HCV classification and typing are important to clinical practice, and the greatest possible range of HCV variants needs to be sought out. The high genetic diversity of HCV is one of the significant problems of future vaccine development. However, only a few local studies had focused on HCV 3a core protein mutations with variable results. Further evaluation is needed to determine the significance of specific mutations in each genotype and how they affect the response to the treatment.
RNA viruses undergo evolution under strong selection pressures, and changes in the epidemiological pattern have been frequently reported. In developing countries like Pakistan, lack of essentials for screening and surveillance on one hand and unsatisfactory health care conditions, on the other hand, are critical factors in the spread of HCV infection. Accurate knowledge of circulating HCV strains using phylogenetic studies and an in-depth understanding of underlying genetic substitutions is, therefore, an important step in identifying epidemiological distribution as well as designing strategies for infection control and drug development. Hepatitis C Virus genotype 3a is more prevalent in Pakistan, and few studies were conducted on complete core protein sequences and phylogenetic analysis in the north-west of Pakistan. Studies performed locally revealed a prevalence of genotype 3a from 26% to 45% (17, 18). According to a national survey conducted in 2007, 4.8% of the Pakistani population was infected with HCV, which corresponds to nearly 9.9 million of overall HCV cases in Pakistan. While the Center for Disease Analysis estimates the presence of 7.1 million HCV cases in the country (19). Changes in the frequency distribution of circulating genotypes of HCV necessitate more research in this regard.
2. Objectives
In the present study, we attempted to amplify, clone, and sequence complete core protein from chronic HCV genotype 3a infected patients of Peshawar, Khyber Pakhtunkhwa province of Pakistan for subsequent phylogenetic analysis. Their sequence analysis and phylogenetic study were performed by comparing them with already reported HCV core protein sequences.
3. Methods
This descriptive study was carried out at the Institute of Basic Medical Sciences, Khyber Medical University Peshawar, Khyber Teaching Hospital Peshawar, Institute of Biotechnology and Genetic Engineering, Agriculture University Peshawar and Center of Applied and Molecular Biology Lahore. The study duration was 24 months (Jan 2014 to Feb 2016). Sera of infected patients was collected in sterile vacutainers and transported to Khyber Medical University for further processing. All procedures were conducted under the declaration of Helsinki.
3.1. Extraction of Viral RNA
Viral RNA was extracted from the sera using an RNA extraction kit (QIAamp® Viral RNA Mini Kit, France) according to the manufacturer’s instructions. RNA was stored immediately at -80ºC for further analysis.
3.2. cDNA Synthesis for Core Gene
cDNA was synthesized by RT-PCR using RT-PCR kit (Invitrogen cDNA protocol kit) according to the manufacturer’s instructions. In brief, 300 ng/µL RNA and 1µL of Outer Antisense (OAS) primer were mixed and incubated at 65ºC for 5 min and kept immediately on ice. Then 4 µL 5x buffer, 2 µL 10 mM dNTP’s, 1µL RNase inhibitor and 1µL reverse transcriptase enzyme (Thermo Fisher Scientific, USA) were added to a final volume of 15 µL. Mixture was kept in a thermal cycler at 42ºC for 60 minutes and then termination of reaction at 70ºC for 10 minutes was carried out.
3.3. Conventional PCR for Core Gene Amplification
For amplification of HCV Core gene, PCR was done in a total reaction volume of 15 µL including 2 µL of cDNA, reverse primer 1 µL, forward primer 1 µL, Master mix (Thermo Fisher Scientific, USA) 7.5 µL and distilled water 3.5 µL. The mixture was kept in the thermal cycler (Techne Inc., USA) and the temperature was set for initial denaturation at 95ºC for 3 min, and then 36 cycles each consisting of denaturation at 95ºC for 30 sec, annealing at 55ºC for 30 sec and elongation at 72ºC for 30 sec. The final extension was carried out for 10 min at 72ºC. Primers were designed using Primer Blast® (20). Forward (5’ATGAGCACACTTCCTAAACC3’) and reverse primers (5’GACGTATTCCGCCACTCTAG3’) specific for core region of HCV were used to amplify cDNA in the thermal cycler (Techne Inc., USA) using Taq DNA polymerase (Fermentas, USA).
3.4. Gel Electrophoresis
The amplified gene-specific PCR product (573 bp) was resolved on 1.5% agarose gel (Thermo Fisher Scientific, USA). It was identified using a 100 bp DNA ladder (Fermentas, USA) and visualized under UV illumination using a gel documentation system (Uvitec Limited, Cambridge, UK). The confirmed PCR product was eluted via (Novel Gel/PCR DNA Purification Mini Kit).
3.5. Cloning and Sequencing of Core gene
In this study, the DH5-α strain of Escherichia coli (provided by Dr. Sami Siraj: Pharmacogenomics Lab KMU) was used to generate chemical competent cell using standard calcium chloride method. Core gene was amplified in a single PCR using gene-specific forward primer (GCGATATCATGAGCACACTTCCTAAA) and reverse primer (AATCTAGATCATGGCTGCTGGATGAAT) with restriction sites for XbaI and EcorV. The amplified product was gel-purified and double-digested using XbaI and EcorV. Similarly, expression vector pDNA3.1/Flag-Tag (Invitrogen, USA) was also double digested using the same enzymes. The resultant products were gel-purified and ligated using T4 DNA ligase kit (Thermo Fisher Scientific, USA). The ligation product was transformed into DH5-α E. coli competent cells through continuous heat shock method. The mixture was then transferred to X-gal-IPTG plates, incubated overnight, and white clones were selected in the next morning. These clones were then propagated in nutrient broth for 18 hours; plasmid DNA was extracted using a standard miniprep method. Extracted DNA was sent to Macrogen (Korea) for sequencing for bi-directional sequencing.
3.6. Alignment and Mutational Analysis
The sequences used in this study were manually curetted by aligning the forward and reverse fragments of the core gene applied to this study. The curetted sequences were used for mutational analysis by aligning these with HCV 3a reference sequence NZL1 accession No D17763.
3.7. Phylogenetic Analysis
Hepatitis C virus complete core gene sequences from different geographical regions of the world were retrieved from the online sequence repositories (NCBI Genbank, DDBJ). Nucleotide sequences (573 bp), as well as the translated amino acid sequences reported from other countries, were aligned using CLUSTAL W of MEGA 6 Software. Sequences retrieved and sequences obtained in this study were used to construct a phylogenetic tree by using the Maximum Likelihood algorithm (ML) of Mega 6 software version 7 (2016, Pennsylvania, USA) (Figure 1).
 with Bootstrap values shown on the branches. The tree shows the phylogenetic relationship of nine newly reported sequences, marked in red with 46 core protein sequences from different geographical regions. The isolate/country/Accession, no of the sequences, are shown here.")
Phylogenetic tree of HCV genotype 3a complete Core protein sequences constructed by ML algorithm (MEGA 6 Software) with Bootstrap values shown on the branches. The tree shows the phylogenetic relationship of nine newly reported sequences, marked in red with 46 core protein sequences from different geographical regions. The isolate/country/Accession, no of the sequences, are shown here.
4. Results
4.1. Nucleotide and Amino Acid Variability in the Core Protein of HCV-3a
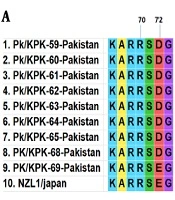
A total of nine HCV core gene clones representing the entire study area were sequenced from the samples. Pairwise nucleotide and deduced amino acid sequences comparison of nine isolates, including PK/59 to PK/65 and PK/68- PK/69, was performed with each other and reference strain (NZL1 Accession No D17763) of HCV 3a genotype. Core protein mutations R70Q (arginine to glutamine) and L/C91M (leucine/cysteine to methionine) were not found among studied isolates. In the sequence of PK/59 at position 72, glutamic acid was replaced with aspartic acid. Position 182 was occupied by leucine in place of phenylalanine in all sequences. Alanine and Serine amino acids at the positions of 189 and 191 were replaced with threonine and cysteine, respectively. In seven of our isolates (PK/59-PK/68), glutamic acid and serine were found mutated to aspartic acid and cysteine, respectively. Amino acid substitutions at various positions are shown in Figure 2.
 aligned against reference 3a sequence (NZL1)")
Amino acid substitutions in core protein sequences (A, position 67 - 73; B, position 90 - 96; C, position 182 - 191) aligned against reference 3a sequence (NZL1)
4.2. Phylogenetic Analysis of HCV Genotype 3a Based on Complete Core Protein Sequences
The HCV 3a core protein sequences was analyzed in Peshawar. The phylogenetic tree was constructed using the Maximum Likelihood (ML) algorithm of Mega software (Mega 6) using the Tamura-Nei model. Nucleotide and deduced amino acid sequences of our nine isolates (accession No.: KY364192, KY434060, MF838728, MF838729, MF838730, MF838731, MF838732, MG977454, MG977455) with HCV 3a isolates from other countries revealed that our sequences shared high homology (100%) (1000 bootstrap replicates). Although our isolates formed a distinct cluster when compared to HCV 3a isolates from other countries, we found that our sequences were clustered together with local sequences in the same geographic area (accession No. HQ108098 and HQ108097) reported earlier from Pakistan. One isolate with accession No. MG-977455 showed homology with previously reported isolate from India (accession No. JQ 717258). The phylogenetic tree of HCV genotype 3a complete core protein sequences constructed by ML algorithm (MEGA 6 Software) is illustrated in Figure 1.
5. Discussion
Currently, classification of HCV is based on the comparison of the nucleotide sequence in different regions of the genome (Core, E1, NS5) or by comparison with available whole genomic sequences or using bootstrap resampling of phylogenetic trees (21). Accurate identification of circulating HCV genotypes is essential for appropriate antiviral therapy. Besides, their epidemiological data can show the migration movement and transmission of infected individuals from an endemic area (22). Phylogenetic relatedness of HCV isolates is a useful tool in understanding the evolutionary history and epidemiology of a virus in a population (23). The rapidly changing epidemiology of HCV disease and its prevalence rates within and between countries is characterized by local variability.
Previous studies have indicated that even within Pakistan, major genotypes of HCV and their prevalence vary considerably (24). As RNA viruses evolve, overtimes we attempted to perform molecular evolutionary and sequence analysis of HCV genotype 3a based on the complete core region of the HCV genome. In this study, complete core protein sequences of HCV genotype 3a were critically evaluated. The analysis of variation in the nucleotide sequence of core protein and its comparison with isolates from other geographical regions is important to understand its global prevalence as well as its management.
In the present study, we observed amino acid polymorphisms at various positions in the HCV core protein. In the HCV genome, mutation may also affect protein protein interactions and response to therapy (25). Previous studies reported that mutations in the core protein are linked with high levels of alpha-fetoprotein (AFP) and liver cancer. Since it is known that there are some relevant core amino acid substitutions associated with increased levels of AFP and subsequent HCC risk. We searched for those mutations in our sequences from the core protein of HCV-3a isolates. Core protein mutations R70Qand L/C91M were not found among studied isolates. Both of these substitutions were more frequent in American isolates compared to Asian ones R70Q (69% versus 26%, P < 0.001) and L/C91M (75% versus 45%, P < 0.001) in patients having HCV genotype 1b infection (14). Previous studies show that R70Q and L/C91M amino acid substitutions in the core protein from different HCV genotypes have been associated with differential responses to antiviral therapy.
Some of these mutations have been found more commonly in viruses infecting patients with a favorable therapeutic response with either complete or partial eradication of the virus in some cases (14). In the PK/59 sequence at position 72, glutamic acid was replaced with aspartic acid. Position 182 was occupied by leucine in place of phenylalanine in all sequences. These substitutions at positions 72 and 182 were also reported by Waheed et al. in 2010. Alanine and serine amino acids at position 189 and 191 were replaced with threonine and cysteine, respectively. In seven of our isolates (PK/59-PK/68), glutamic acid and serine were found mutated to aspartic acid and cysteine, respectively. These substitutions of core protein do not affect encapsidation. Further studies are required to evaluate the effect of various amino acid substitutions on core protein functions and to analyze their impact on the clinical outcome of this disease.
Although changes in the epidemiological pattern of HCV infection have been previously reported from different provinces of Pakistan, still HCV genotype 3a is the most frequently reported subtype (58%) in Peshawar (26). In the present study, genetic relatedness with local isolates points to the prior presence of similar types in other parts of Pakistan. Recently, Gul et al. (17) described the evolutionary analysis of HCV genotype 3a isolates from Peshawar. Based on their study findings, HCV genotype 3a isolates clustered closely with both local isolates and with HCV 3a isolates from neighboring countries, including Iran and India, as HCV 3a genotype is also prevalent in these regions (17). Consistent with their findings, HCV 3a isolates analyzed in the current study demonstrated high homology with previously analyzed genotype 3a isolates. A study conducted by Waheed et al. (25), using phylogenetic analysis showed that HCV gene sequences were clustered with sequences from India; however, the same group in 2010 performed phylogenetic analysis of entire core gene of HCV 3a genotype from Pakistan and found a close association with HCV genotype 3a reported from Japan (12).
Such a difference in the clustering patterns of different HCV genes is potentially due to mutation and sequence variations. The diversity in the clustering pattern of HCV genotypes could be attributed to the continuously evolving nature of RNA viruses. This high genetic diversity is the result of error-prone HCV polymerase enzyme, which lacks the ability of proofreading with 10-3 10-5 mutations per genomic replication. Genomic diversity is associated with two important parameters: the half-life of HCV is extremely short, and HCV polymerase lacks the proofreading ability. An important observation regarding the origin of HCV 3a genotype in Pakistan was made based on study of the complete genome of HCV genotype 3a. Ghori et al. in 2016, showed that Pakistani sequences share a common ancestor and are more closely related to each other than to sequences from other countries (26). It has been observed that epidemiological parameters, especially changes in the disease transmission patterns, play an essential role in the transmission of prevalent genotypes in a population. In particular, the migration of people within a country might be responsible for the spread of prevalent infections.
5.1. Conclusions
This study reports HCV complete core protein sequences from HCV 3a-infected persons of Peshawar along with phylogenetic analysis. Core protein amino acid sequences could be utilized as a useful phylogenetic marker for establishing evolutionary relationships among circulating viral strains. Phylogenetic analysis of HCV 3a genotype revealed that the obtained sequence of core protein has high homology with other isolates previously reported from Pakistan. Owing to its prevalence in Pakistan, the evolutionary study of HCV genotypes will be helpful in the development of vaccine and antiviral treatment. In addition, further studies of HCV 3a genotype based on complete genome analysis are required for a better understanding of evolutionary aspects of HCV genotypes prevalent in Pakistan.
