






1. Background
Seasonal influenza poses a significant global threat, leading to disease and mortality by causing acute respiratory illness in humans (1, 2). Annually, influenza epidemics manifest through subtypes H3N2 or H1N1 (3). The third pandemic of influenza in the 20th century was initiated in 1968 with the first documented influenza A/H3N2 outbreak.
Influenza viruses, classified under the orthomyxoviridae family, possess a genome comprising eight single-stranded RNAs with a negative sense (4, 5), which are categorized into 3 varieties (A - C), with type A posing the most significant risk to human health (6).
The surface glycoproteins hemagglutinin (HA) and neuraminidase (NA) are pivotal targets for the host immune system, distinguishing between influenza A virus subtypes. Hemagglutinin surface proteins facilitate virus attachment to sialic acid receptors, enabling virus entry through endocytosis and subsequent fusion. The NA surface protein, acting as a receptor-disrupting enzyme, plays a crucial role in viral release and cell-to-cell propagation. The influenza virus HA protein undergoes cleavage into HA1 and HA2 components through the action of the host cell protease enzyme.
The HA1 subunit features antigenic sites, specifically epitopes A, B, C, D, and E (7, 8). Amino acid (AA) alterations on the surface of HA and NA induce antigenic drift, reducing identification by host immune responses (6). Strains utilized in influenza vaccinations require frequent updates due to antigenic drift (9, 10). Recent identifications include 18 HA and 11 NA subtypes (7, 11).
Common antigenic drift variants appear in H3 of H3N2 viruses (12). Seasonal influenza A/H3N2 viruses continually evolve through antigenic drift in the HA surface glycoprotein, evading existing immunity in the human population. Characterizing genetic and antigenic changes resulting from this drift is crucial for selecting optimal vaccine strains and assessing vaccine effectiveness against emerging variants. The HA head domain encompasses five antigenic sites, with critical residues within these sites playing a significant role in antigenic change. Phylogenetic analysis of HA sequences can also identify the emergence of new genetic clades and subclades that may become predominant (13, 14).
In recent years, influenza A/H3N2 viruses have diversified into multiple genetic clades and subclades, with clade 3C viruses currently predominant globally. However, the distribution and evolution of these subclades can vary by geographic region. Ongoing genetic and antigenic surveillance is essential to monitor mutations affecting antigenicity, especially in regions with limited data.
2. Objectives
This study aims to elucidate the molecular variations and phylogenetic relationships of locally circulating influenza A/H3N2 strains through genetic analysis of the critical HA gene. Detecting amino acid substitutions, particularly in vital antigenic sites, and tracking the circulation of genetic clades/subclades provide crucial information for vaccine strain selection and preparedness for future epidemics.
This analysis offers a vital baseline characterization of recently circulating strains for assessing future antigenic drift (13, 15, 16). Seven H3N2 clades (labeled clades 1 to 7) and numerous subclades have recently emerged. Class 3 viruses are currently the most common, with three primary subclasses, 3A, 3B, and 3C, comprising various distinguishable gene groups (17, 18). Subclass 3C.2a viruses are endemic in numerous regions worldwide, while subclass 3C.3a viruses are endemic in Asia, Eastern Europe, and Africa (19).
Global influenza virus surveillance is indispensable for identifying variants such as vaccine escape variants and antigenic shift viruses. Consequently, this study aims to assess the molecular characterization and phylogenetic analysis of the influenza A/H3N2 HA gene.
3. Methods
3.1. Subjects
This study was conducted on 300 nasopharyngeal swab samples, randomly collected from patients admitted to Shiraz, southern Iran’s clinics during the 2015 - 2016 influenza season (from October 2015 to February 2016). The inclusion criteria were patients presenting with fever (> 38°C) and respiratory symptoms such as cough, sore throat, and runny nose. The randomization process employed simple random sampling methods.
Nasopharyngeal swabs collected samples by inserting a sterile swab through the nostril to the nasopharynx. Subsequently, the swabs were placed in transport media and then transported to the virology laboratory of Shiraz University of Medical Sciences. The samples were meticulously stored there at -80°C for further examination.
Informed consent was acquired from individuals by the guidelines of the Research Ethics Committee of Shiraz Medical University (IR.SUMS.MED.REC.1396.S269) prior to obtaining samples from each patient.
3.2. RNA Extraction
Total RNA extraction from clinical samples followed the manufacturer's instructions, employing a high-purity viral RNA kit (Roche, Product No.: 11858882001). Subsequently, electrophoresis on a 1% (w/v) agarose gel was used to assess the integrity of the total RNA. Spectrophotometry confirmed the extracted RNA's purity and quality ratio of 1.8 to 2. The isolated RNA was then preserved at -80°C before further processing.
3.3. Real-time PCR
Real-time polymerase chain reaction (PCR) experiments were conducted to detect influenza and H3N2 viruses. The primers and probes for this experiment, listed in Table 1, were produced by the Metabion Company. A probe mentioned earlier was utilized, adhering to World Health Organization (WHO) guidelines (20).
For the real-time PCR experiment, the SuperScriptTM III Platinum® One-Step real-time PCR System was employed with Platinum Taq DNA Polymerase from Invitrogen (lot number: 1312837) to create the master mix. The Rotor-Gene 6000 (QIAGEN) real-time PCR machine facilitated the procedure. The experiment included 50ng of RNA, 0.4 µL of each primer (0.4 µM), 0.4µl of the probe (0.2 µM), 12.5µl of 2X Reaction Mix, and 0.4 µL of Superscript III RT/Platinum ready-to-use Taq, resulting in a 25 µL reaction. Adequate nuclease-free water was added to ensure a final volume of 25 µL for each reaction.
| Primers and Probes | Sequence (5’-3’) | Working Concentration (μM) |
|---|---|---|
| InfA Forward | GACCRATCCTGTCACCTCTGAC | 40 |
| InfA Reverse | AGGGCATTYTGGACAAAKCGTCTA | 40 |
| H3 Forward | TAGGATCCAATCTTTGTATCCTGACTT | 40 |
| H3 Reverse | GATGTGTACAGAGATGAAGCATTAAACA | 40 |
| InfA Probe | Fam-TGCAGTCCTCGCTCACTGGGCACG-BhQ | 10 |
| H3 Probe | Fam-AGCTCAACACCTTTGATCTGGAACCGG-BhQ | 10 |
The Primers and Probes Utilized in Real‐time PCR Assay
3.4. Full-length Amplification of HA Segments
The amplification of the HA gene was conducted through a Qiagen One-step Reverse transcriptase polymerase chain reaction (RT-PCR) Kit (Qiagen, Hilden, Germany), utilizing the primers specified in Table 2. Given the relatively lengthy sequence of the A/H3N2-HA gene, two segment-specific primer sets were devised for amplification (Table 2).
| Influenza Subtype | Primer Sequence (5′-3′) | Target | Tm | PCR Product (bp) |
|---|---|---|---|---|
| H3N2 | F: GAGCTACATTCTATGTCTGG, R: CTTAACATATCTGGGACAGG | HA1 | 55 | 958 |
| H3N2 | F: CATCACTCCAAATGGAAGCA, R: TGTTGCACCTAATGTTGCC | HA1 | 55 | 800 |
Primer Sets Utilized in One-step RT-PCR to Amplify the Coding Area of the HA Gene of the A/H3N2 Subtypes Along with Their Sequences and Binding Sites
The one-step RT-PCR was executed in a 20 µL total volume, comprising 2 × 10 µL one-step reaction mix, 0.4 µL forward and reverse primers, 0.4 µL one-step enzyme mix, 2 µL template (RNA), and 6.8µL RNase-free water. The thermal cycler program was initiated after the PCR mix preparation, as outlined in Table 3. The PCR products underwent further purification using the GF-1 PCR cleanup kit post-examination on a 1.5% agarose gel (Vivantis, Malaysia).
| PCR Steps | Temperature (°C) | Time | Cycles No. |
|---|---|---|---|
| cDNA synthesis | 45 | 30 min | 1 |
| Denaturation | 95 | 5 min | |
| Secondary denaturation | 95 | 30 s | 35 |
| Annealing | 55 | 30 s | |
| Elongation | 72 | 2 min | |
| Final elongation | 72 | 10 min | 1 |
| Hold | 4 | - |
One-step RT-PCR Temperature Protocol for HA Gene Amplification
At Sequence Laboratories, First Base Company, Malaysia, the purified DNA underwent genetic analysis employing an ABI sequence genetic analyzer (Applied Biosystems, Foster City, Calif.). Sequences were modified and compiled using CLC Sequence Viewer V.6.7 and Chromas Lite version 2.6.1 (Technelysium Pty Ltd., Australia). The nucleotide sequences of all samples were submitted to the GenBank database, and the corresponding accession numbers are detailed in Table 4.
| Subtype | Strain | Collection Date (DD/MM/YYYY) | Accession Number (HA Gene) |
|---|---|---|---|
| A/H3N2 | A/Shiraz/1002/2015 | 02/10/2015 | MG021157 |
| A/Shiraz/1003/2015 | 05/10/2015 | MG021153 | |
| A/Shiraz/1004/2015 | 07/10/2015 | MF993042 | |
| A/Shiraz/1007/2015 | 02/11/2015 | MF993040 | |
| A/Shiraz/1008/2015 | 09/11/2015 | MF993041 | |
| A/Shiraz/1009/2015 | 13/11/2015 | MG021158 | |
| A/Shiraz/1010/2015 | 13/11/2015 | MG021155 | |
| A/Shiraz/1011/2015 | 22/11/2015 | MG021152 | |
| A/Shiraz/1012/2015 | 28/11/2015 | MG021159 | |
| A/Shiraz/1013/2015 | 03/12/2015 | MG021154 | |
| A/Shiraz/1014/2015 | 10/12/2015 | MF164913 | |
| A/Shiraz/1015/2015 | 10/12/2015 | MF993043 | |
| A/Shiraz/1016/2015 | 20/12/2015 | MF993044 | |
| A/Shiraz/1017/2015 | 25/12/2015 | MF993045 | |
| A/Shiraz/1018/2015 | 25/12/2015 | MF993046 | |
| A/Shiraz/1019/2016 | 02/01/2016 | MG021156 | |
| A/Shiraz/1020/2016 | 08/01/2016 | MG021160 | |
| A/Shiraz/1021/2016 | 17/01/2016 | MG021161 | |
| A/Shiraz/1022/2016 | 24/01/2016 | MF993047 | |
| A/Shiraz/1023/2016 | 02/02/2016 | MF993048 | |
| A/Shiraz/1024/2016 | 05/02/2016 | MG021162 | |
| A/Shiraz/1025/2016 | 10/02/2016 | MG021163 |
The Accession Number of Influenza A/H3N2 Isolates Registered in the GenBank Database
3.5. Multiple Sequence Alignment and Phylogenetic Analysis
The National Center for Biotechnology Information's GenBank served as the reference for IAV-HA nucleotide sequences. All HA gene sequences were aligned using the Clustal W alignment tool within the Bioedit program version 7.2.5. Based on the MEGA 7.0 implementation of the best-fit nucleotide substitution model for the HA gene (12), the maximum likelihood was employed to construct the phylogenetic tree (21). Bootstrap scores exceeding 75% were considered significant after 1000 bootstrap repeats, assessing the tree's reliability. The HA phylogenetic clustering pattern was employed to designate clades.
4. Results
4.1. Demographic Data and Patient Characteristics
On average, patients were 35 ± 2.40 years old, with 193 males and 107 females. Real-time PCR analysis identified 52 (17.33%) swab samples from influenza-like illness (ILI) patients as positive for influenza A, and among these, 22 were influenza A/H3N2 positive, consisting of 13 males and nine females.
4.2. Sequence Analysis
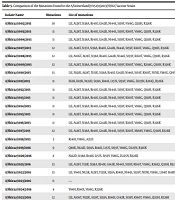
The entire HA gene sequences of 22 H3N2 samples from Shiraz, Iran, were examined using the A/Switzerland/9715293/2013 (H3N2) vaccine strain as a reference. At the AA level, the identity percentage ranged from 98.37% to 99.45%. Except for one strain, A/Shiraz/1023/2016, all 22 H3 protein sequences exhibited mutations compared to A/Switzerland/9715293/2013 (H3N2), with the highest number of mutations observed in A/Shiraz/1010/2015 and A/Shiraz/1022/2016 (15 mutations each).
A comparative analysis with A/Switzerland/9715293/2013 (H3N2) observed an average of 6 (range 0 - 9) substitutions at all antigenic sites. Nineteen (86%) isolates shared four mutations at the AA antigenic site, including S138A and R140I at site A and S159Y and V186G at site B. The R140I and V186G substitutions were predominant mutations in all strains except A/Shiraz/1023/2016. One strain (A/Shiraz/1011/2015) exhibited a unique substitution, K83R, at antigenic site E.
Other alterations at antigenic sites included N121K, N122D, T131K, T135K, S137A, and N144S at site A; K160T, Q197K, S198P at site B; D225N at site D; and Y94H at site E. Antigenic site C, which did not show any mutations. Additionally, substitutions outside the antigenic locations were observed, as detailed in Table 5.
| Isolate Name | Mutations | List of Mutations |
|---|---|---|
| A/Shiraz/1002/2015 | 10 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, V186G, Q311H, R326K |
| A/Shiraz/1003/2015 | 11 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1004/2015 | 11 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1007/2015 | 12 | L3I, A128T, S137A, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1008/2015 | 12 | L3I, A106T, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1009/2015 | 12 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, R208G, Q311H, R326K |
| A/Shiraz/1010/2015 | 15 | L3I, N121K, A128T, T135K, S138A, R140I, G142R, N144S, S159Y, K160T, N171K, V186G, Q197K, Q311H, R326K |
| A/Shiraz/1011/2015 | 11 | E62K, K83R, N122D, S138A, R140I, L157S, S159Y, V186G, D225N, R261Q, R326K |
| A/Shiraz/1012/2015 | 11 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1013/2015 | 11 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1014/2015 | 11 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1015/2015 | 11 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1016/2015 | 11 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, Q311H, R326K |
| A/Shiraz/1017/2015 | 12 | L3I, A128T, S138A, R140I, G142R, N144S, S159Y, K160T, M168V, V186G, Q311H, R326K |
| A/Shiraz/1018/2015 | 3 | R140I, V186G, A212S |
| A/shiraz/1019/2016 | 9 | Q80K, N122D, S138A, R140I, L157S, S159Y, V186G, D225N, R326K |
| A/Shiraz/1020/2016 | 8 | N122D, S138A, R140I, L157S, S159Y, V186G, D225N, R326K |
| A/Shiraz/1021/2016 | 13 | L3I, A128T, T131K, S138A, R140I, G142R, N144S, S159Y, K160T, V186G, R261Q, Q311H, R326K |
| A/Shiraz/1022/2016 | 15 | L3I, Y94H, N121K, A128T, T135K, S138A, R140I, N144S, S159Y, N171K, V186G, L194P, S198P, Q311H, R326K |
| A/Shiraz/1023/2016 | 0 | ----- |
| A/Shiraz/1024/2016 | 4 | Y94H, R140I, V186G, R326K |
| A/Shiraz/1025/2016 | 12 | L3I, A106T, N121K, A128T, S138A, R140I, G142R, S159Y, K160T, V186G, Q311H, R326K |
Comparison of the Mutations Found to the A/Switzerland/9715293/2013(H3N2) Vaccine Strain
4.3. Phylogenetic Tree Analysis
The phylogenetic tree analysis revealed the presence of sub-clades 3C.2a and 3C.3 in comparing the northern hemisphere vaccination strain (A/Switzerland/9715293/2013(H3N2)) to the A/H3N2 strain for the season (Figure 1). Notably, the majority of strains (16; 72.72%) belonged to sub-clade 3C.2a, while an additional six isolates (27.27%) grouped with sub-clade 3C.3 viruses and represented by A/Hong Kong/4801/2014 and A/Switzerland/9715293/2013, respectively.
A/Shiraz/1023/2016 (H3N2) among the isolates exhibited a striking similarity, clustering closely within subclade 3C.3. No mutations were observed when compared to A/Switzerland/9715293/2013 (H3N2), emphasizing its close relationship within this specific genetic context.

Phylogenetic analysis of the HA1 sequence
5. Discussion
Conducting thorough surveillance of influenza viruses during the early phases of influenza season is crucial for detecting prevalent strains and designing effective vaccines. Mainly, influenza A/H3N2 exhibits rapid mutational dynamics, necessitating vigilant detection efforts. The observed mutations in the antigenic sites of Shiraz samples suggest a discernible drift in the HA gene of the H3N2 virus.
Antigenic evolution raises concerns about potential resistance to vaccine strains selected by the WHO, potentially compromising immunity to emerging virus strains provided by existing vaccines. Hence, characterizing early-season influenza viruses becomes imperative for informed decisions in formulating forthcoming influenza vaccines.
Evidence suggests a higher propensity for AA substitutions in HA proteins of influenza A/H3N2 than A/H1N1 (22, 23). Furthermore, notable shifts, such as the dominance of subclade 3C.2a in the northern hemisphere, have been documented in influenza studies (24, 25).
According to the comparison of HA coding sequences with WHO reference strains, subclade 3C.2a viruses exhibited global prevalence, whereas subclade 3C.3a viruses were prevalent in Asia, Eastern Europe, and Africa (19). In addition, 72.72% of our isolates were from subclade 3C.2a. At the same time, six out of 22 belonged to clade 3C.3. Notably, a prior study in Shiraz highlighted a prevalence of subclade 3C.3b (26), where most viruses were within clade 3C.2, with one isolate falling under 3C.2a.
Comparisons with studies conducted by Monamele GC and Korsun N yielded similar results (11, 27). The sequence analysis of our data aligned with previous seasons in Shiraz and mirrored global trends, consistent with WHO data from the Northern Hemisphere flu season (26, 28).
The phylogenetic analysis, indicating the isolates clustering in subclades 3C.2a and 3C.3a, reflects global trends and hints at the localized evolution of H3N2 viruses distinct from the predominant global strains. Hence, ongoing genetic characterization is essential to monitor novel variants and gain insight into the intricate dynamics of H3N2 influenza strains.
Critical viral processes like glycosylation and deglycosylation play pivotal roles in concealing antigenic epitopes, evading humoral immune responses, and altering receptor binding affinity (29, 30). There is a high risk of antigenic drift in the gene that encodes the HA surface glycoprotein, which is the target of neutralizing antibodies. Five neutralizing antigenic sites in influenza A/H3N2 are denoted by letters A through E, with 131 AA locations linked to antigenic variation (31). Notably, seven of these positions are concentrated in antigenic sites A and B, specifically at positions 145, 155, 156, 158, 159, 189, and 193 (31, 32). Substitutions occurring at these key positions and antigenic sites are crucial. However, this study revealed numerous alternatives, only one (S159Y) localized to these vital positions, potentially facilitating immune evasion. An amino acid alteration at position 159 of H3N2 influenza strains circulating in our region may indicate antigenic drift due to its location in a highly exposed region of the B antigenic site (27). Vaccine protection is significantly diminished by mismatches between circulating strains and vaccines, resulting from antigenic drift.
Epidemiologically significant antigenic drift variations typically necessitate at least four changes co-occurring at two or more antigenic sites (12, 27, 33, 34). Studies by Korsun et al. in Bulgaria identified four substitutions at A and B antigenic sites (27), while previous research in Iran revealed 5 substitutions - R142G, N145S, T128A, P198S, and S219F (26). Results from Cameroon during the flu season indicated substitutions A138S, F159Y, and K160T (11), aligning with findings from Germany and Iran involving mutations at 186 and 198 sites, respectively (18, 26). The observation that only one substitution occurred at the 7 crucial antigenic residues despite multiple mutations in antigenic sites suggests that major antigenic changes have not yet occurred. Nevertheless, the accumulation of substitutions near these critical antigenic sites hints at the potential for future antigenic drift.
The results showed that 22 (7.33%) were influenza A/H3N2 positive. Moasser et al. reported that in Iran, 36 (18%) out of 200 samples were A/H3N2 (26), and in Bulgaria, 10% of the isolates were influenza A/H3N2 viruses (27). In addition, 193 males and 107 females were among the 300 patients, of whom 22 were influenza A/H3N2-positive subjects, 13 were males, and nine were females. In addition, patients' ages ranged from 3 to 19, with a mean age of 35 ± 2.40 years. Korsun et al. studied patients from 13 days of age to 88 years old, of whom 676 (48.7%) were male (27).
A limitation of this study was that the relatively small sample size from a single season likely did not capture the full diversity and antigenic drift of regional H3N2 viruses, underscoring the need for larger multi-season studies. Additionally, functional analyses of the impacts of the observed mutations on antigenicity using serological assays were not performed but are warranted in future work to better characterize the potential antigenic drift.
In conclusion, the genetic variations of influenza A/H3N2 indicate that new variations may develop and evade the immune system, invalidating vaccination. Based on the results, it is necessary to continue molecular research on the virus for efficient influenza management and to support the genetic variability of influenza A/H3N2. Ongoing molecular surveillance through genomic sequencing of regional influenza viruses, especially vaccine strains like H3N2, is critical for continued monitoring of viral evolution and the emergence of new variants. Additionally, antigenic characterization of the current and future viral isolates using serological assays and epidemiological analyses evaluating the relationships between genetic changes and vaccine effectiveness would provide important insights into the functional and clinical significance of the observed mutations.
