



1. Introduction
Sjogren-Larsson syndrome is a rare autosomal recessive neurocutaneous disorder characterized by an inborn lipid metabolism error resulting from a mutation in the ALDH3A2 gene. This gene encodes the enzyme fatty aldehyde dehydrogenase (FALDH), and its mutation results in a decrease in the amount of FALDH present in the body, leading to a deficit in the oxidation of the long chain of fatty acids, generating a deposit of lipid metabolites in the tissues (1, 2). Deposits in the skin and central nervous system (CNS) are primarily manifested through the classic triad of congenital ichthyosis, paraplegia or quadriplegia, and mental retardation (1-3). Ichthyosis results from the disorganization and disruption of the transepidermal water barrier by the agglutination of lipid metabolites in this region, while CNS manifestations originate from disorders of myelination and demyelination of nerve fibers due to the deficiency of oxidized fatty acids (2, 4, 5). Ocular and visual changes are also frequent and result from deposits of lipofuscin pigment in the retinal epithelium (2, 6). Most reported cases have serious repercussions (1).
2. Case Presentation
We report the cases of two patients, one female and one male, aged three months, triplets from consanguineous parents (third cousins), premature at 35 weeks, presenting similar clinical conditions at birth, while the third sister did not present with cutaneous or systemic manifestations. They exhibited erythematous and xerotic skin at birth, evolving shortly afterward with apnea episodes, cyanosis, dysphagia, vomiting, and malnutrition. One patient, the girl, experienced seizure episodes. The other patient, the boy, had spasticity.
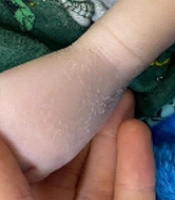
Dermatological examination revealed diffuse xerosis of the skin in both patients (Figure 1), with fine scaling and predominance on the face; an erythematous macula extending from the glabella to the right upper eyelid; a small scaly erythematous plaque in the right cervical region in the girl (Figure 2); and discolored skin, diffuse cutaneous xerosis, with fine scaling, predominantly on the face, distal third of the lower limbs and feet in the boy (Figure 3). They presented with paronychia, nail hyperkeratosis, detachment, and dystrophy of the nail plate in the nails of the hands, with no changes in the toenails.

Diffuse cutaneous xerosis. Emphasis on the abdominal region

Xeroderma on the face and scaly erythematosis lesions in the glabellar region. Erythematous scaly plaque in the neck region

Xeroderma on the face. Fine scaling on the dorsum of the left foot
Together with other specialties, the diagnostic hypotheses of inborn error of metabolism, Otahara epileptic syndrome, congenital ichthyosiform erythroderma, and Sjögren-Larsson syndrome were proposed. Sjögren-Larsson syndrome was considered because of the systemic picture presented by the patients, the parents' consanguinity, and skin biopsies performed in the initial evaluation, revealing hyperkeratosis, accentuation of the granular layer and atrophy of the epidermis, with preservation of the dermis and subcutaneous tissue, compatible with ichthyosis.
Nail alterations in this syndrome have been poorly described in the literature. The acute changes observed were possibly due to inflammatory processes and metabolic stress during hospitalization.
In the reported cases, intensive skin hydration was used, which resulted in an improvement in the dermatological condition, despite serious systemic repercussions.
3. Discussion
Diffuse erythema at birth characterizes the onset of the dermatological manifestations of this syndrome, which essentially evolves with lamellar ichthyosis due to a defect in keratinization associated with pruritus. Classically, the folds, periumbilical, and neck regions are affected, while the face is generally spared, which differs from the cases presented here. Palmoplantar keratoderma can also be seen (1, 2, 7). Neurological involvement is demonstrated by progressive delay in motor development, pyramidal syndrome, and spasticity. It is possible that spasticity, in more advanced stages, affects the upper limbs and perioral region, causing speech and swallowing difficulties. Seizures, as presented by the patient in this case, are common and present in up to 40% of the cases. Premature birth has been commonly reported by researchers, as also observed in our case (7-9). The diagnosis is obtained through the presence of the classic triad and changes in the eye fundus, with crystalline maculopathy observed in up to 100% of the cases (2). Confirmation was made by enzymatic analysis, with the demonstration of FALDH deficiency in fibroblast or leukocyte cultures, which was not available in our service. It should be noted that only half of the patients with neurological symptoms similar to this syndrome, together with skin changes, have a fatty aldehyde dehydrogenase deficiency. Evaluating the urinary excretion of leukotriene B4 and its metabolites and genetic studies of the ALDH3A2 gene can help diagnose; however, it is not essential for confirmation. These tests should be performed whenever possible for diagnostic confirmation and genetic counseling (1-4, 7). Furthermore, it is necessary to make a differential diagnosis from other forms of ichthyosis, such as X-linked recessive, vulgaris, lamellar, bullous, and non-bullous congenital ichthyosiform erythroderma. The treatment is multidisciplinary, and dermatological treatment involves the topical use of emollients, keratolytics, and, in some cases, the use of systemic retinoids (1-4, 7).
We present this case due to its importance, and this description is extremely valid mainly because of the rarity of the presentation, especially because these are triplet patients in which two of the twins have the syndrome and one of the twins does not. In addition, it serves as an alert to physicians for correct investigation, correct diagnosis, and multidisciplinary and dermatological follow-up.
