


1. Introduction
Kaposiform haemangioendothelioma (KHE) is a rare vascular tumor, occurring predominantly in infancy, with an incidence of 0.091 per 100,000 children. Typically presenting as a solitary large cutaneous lesion with no distant metastases. Children with KHE are at high risk of developing the Kasabach-Merritt phenomenon (KMP), a consumptive coagulopathy. KMP is defined as profound thrombocytopenia, hypofibrinogenemia, and elevated D-dimer. KMP has a mortality of 10 - 30%. Due to its rarity, there is little robust evidence for KHE management; treatment guidelines for KHE are based mainly on case reports and expert consensus, with few clinical trials. Rapamycin (mTOR) inhibitors are a safe and effective treatment.
2. Case Presentation
A full-term female neonate was born by vaginal birth at 39 + 1 weeks. It was hemodynamically stable, afebrile, and had a normal APGAR score at birth. The pregnancy and antenatal investigations were unremarkable.
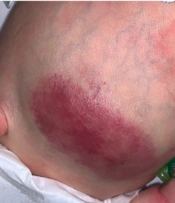
An indurated non-ulcerated violaceous plaque of 7 × 4 cm size (Figure 1) across the abdomen was noted at birth.

KHE on day 2 of life
2.1. Clinical Findings and Assessment
On day 2 of life, the lesion had enlarged (7.5 × 4.5 cm) with increasing induration. There were signs of a consumptive coagulopathic process, low fibrinogen (1.65g/L [reference range 2.70 - 2.83 g/L]), elevated D-dimer (5.49 mg/L), and thrombocytopenia (platelet count 110 × 109/L [reference range 150 - 500 × 109/L]).
Abdominal ultrasound found a hyper-vascular mass in subcutaneous tissue, depth 11mm, of the anterior abdominal wall compatible with a vascular malformation. MRI revealed a complex heterogeneous left abdominal subcutaneous tissue lesion (17 × 55 × 58 mm), with increased T2 signal and internal flow voids, consistent with a large vascular malformation. A punch biopsy under general anesthetic was performed, with differentials of tufted angioma or KHE.
The initial stain was non-diagnostic, with discreet lobulated proliferations within the dermis and subcutaneous fat. On higher power, spindle cells coalesced to form slit-like spaces, no cytological atypia of the spindle cells, and mitosis was rare.
Immunohistochemistry staining showed endothelial cells were strongly and diffusely reactive for CD34 (Figure 2) and within the lobular proliferation for ERG and CD31. D2-40 demonstrated a sweeping pattern of spindle cells around smaller nodules of endothelial cells, and CD61 highlighted amorphic fibrin thrombi, both characteristic of KHE (1). A multidisciplinary team assessed the presentation, diagnosing KHE with KMP. Excision and embolization were deemed not possible due to size and vascularity.

The CD34 marker was strongly and diffusely positive
2.2. Intervention and Outcome
She was commenced on oral prednisolone 1 mg/kg/day on day 6 of life, with initiation of sirolimus on day 16 of life at 0.8 mg/m2/daily. Target trough sirolimus levels at 5 - 15 µg/L (2).
4-days after commencing sirolimus, the lesion had grown to 5 × 9 cm. There was worsening consumptive coagulopathy, platelet count 16 × 109/L, the nadir, with decreasing fibrinogen (1.12 g/L), rising D-Dimer (7.92 mg/L), sirolimus trough level was 2.3 µg/L. Sirolimus and prednisolone increased to 0.8 mg/m2/twice daily and 2mg/kg/daily, respectively. On day 24 of life, the lesion was softer, with reduced bruising, sirolimus in the target range, and a rising platelet count. This trend continued; prednisolone was weaned from day 29 of life with continued weight-based sirolimus.
Staging ultrasounds at 12 and 14 months old showed minimal shrinkage, suggesting sirolimus was unlikely to be of further benefit. There was minimal vascularity of the tumor remaining, and surgical resection with a primary closure was performed with a desire to cease medication. Pathology showed residual involvement at the margins of the resection. No significant surgical complications. The patient continued sirolimus; dose wean commenced at 15 months old, lowering to 0.1 mg fortnightly, with cessation at 17 months old with close clinical monitoring for recurrence. To date, there is no recurrence at 21 months of age.
3. Discussion
The presentation of a rapidly progressing KHE lesion and KMP in infancy presented a treatment dilemma. In a large retrospective review of 107 patients (3), truncal KHE is uncommon in 20% of presentations; however, these lesions are high-risk for KMP, with 75% development (3).
Diagnosis of KHE is complex and requires a correlation of clinical, radiological, and histological elements. KHE has a different treatment algorithm than other vascular anomalies; thus, a firm diagnosis is vital (1). Ultrasound, which is accessible and non-invasive, is the initial modality of choice, and MRI has a role in diagnosis and assessing the extent of the lesion (1).
Biopsy is the gold standard for diagnosis but can be challenging with severe KMP-related thrombocytopenia (1). Histologically, KHE has a hallmark of infiltrating, defined, rounded, confluent nodules with spindle cells (1), which form slit-like vascular lumina and malformed lymphatic channels. Immunohistochemistry staining of KHE endothelial cells is positive for vascular endothelial markers (CD31, CD34), D2-40, VEGFR-3, ERG, and negative for Glut-1 and HHV-8 staining (1, 4).
KHE treatment guidelines are based mainly on case reports and expert consensus, with few clinical trials. Rapamycin inhibitors have recently developed a large evidence base for treatment in KHE (1, 2), with reduction in KHE size and normalization of platelet counts, in combination with corticosteroids, are considered the recommended treatment of KHE with KMP (1). The treatment regimen for sirolimus has not been established, and though target ranges reported in the literature have varied (1, 2), the most common is 5 - 10 µg/L (2). Little is published in the literature surrounding risk factors for adverse effects of sirolimus for KHE. A systematic review of the use of sirolimus in children with lymphatic malformations did not find a correlation between adverse events and sirolimus levels (5). Duration of therapy is another undefined area, ranging from 6 - 24+ months in the literature and reports of recurrence once the medication is ceased (1, 2, 5). Corticosteroids are not recommended as monotherapy, given low response rates of 10 - 27% (1).
Vincristine monotherapy does not appear to be effective for KHE patients (1). It was recommended as first-line therapy in guidelines prior to the publication of large sirolimus studies (6). Additionally, vincristine requires central venous access and in-patient administration (1).
Surgical resection is considered potentially curative and could be considered early in patients where it can be done safely with KHE of appropriate size and stability (1, 6, 7). However, it is not commonly performed due to the vascularity of the lesions, the commonly associated coagulopathy, the typically young age of the patients, and concern that invasive interventions will worsen the hematological parameters (6). Few case reports (7, 8) detail surgical excision following response to systemic treatment with excellent outcomes and no recurrence. No longitudinal studies are looking at outcomes in patients surgically managed. Given the long-term safety of sirolimus in young children is not well-documented and reports of recurrence once ceased (9), surgical excision should be considered when safe. In a small case series, there was no recurrence of KHE following surgical resection (7), but there are case reports of late-term recurrence post-surgical intervention (8). This study demonstrates clinicians' difficulties in managing extremely young patients with complex, rare diseases with high mortality risk. A systemic, multidisciplinary approach, including an uncommon but successful surgical intervention, was successfully utilized. A limitation of the approach to this case study is the single patient and relatively short duration of follow-up.
3.1. Conclusion
This case highlights the complexities of diagnosing a rare illness in a patient with a rapidly progressive disease. Our patient responded excellently to a combination of sirolimus, prednisolone, and surgical treatment, although follow-up will continue long-term, given the involvement of surgical margins. There is a lack of consensus-driven guidelines on optimal sirolimus dose and duration, the decision and timing of surgery, complicating management decisions for clinicians. Further research is needed to assess treatment regimens and the long-term prognosis of patients with KHE.
