


1. Context
Rheumatological disorders are considered rare in children, with a global incidence of 2% - 5% (1). However, this number may be higher in developing countries due to underreporting. Approximately 10% - 20% of all rheumatological disorders in adults present initially during pediatric age groups (2).
Rheumatological diseases exhibit a wide variety of clinical presentations involving the skin, musculoskeletal system, vasculature, and various organ systems. Dermatologic features may be the only presenting features in some rheumatological diseases.
Common rheumatological diseases in children include acute rheumatic fever (ARF), Kawasaki disease (KD), Henoch-Schönlein purpura (HSP), systemic lupus erythematosus (SLE), systemic-onset juvenile idiopathic arthritis, auto-inflammatory syndromes, juvenile dermatomyositis, juvenile scleroderma, juvenile psoriasis, periodic fevers, and many others. Early recognition of these rashes aids in the diagnosis of underlying rheumatological diseases and initiating treatment. Medications may also lead to dermatological manifestations and are implicated in drug-induced lupus. Rheumatological diseases may have a genetic basis, but the dermatological manifestations represent a wide spectrum of disease presentations. Inflammatory bowel disease (IBD), diabetes mellitus, psoriasis, seronegative enthesopathy and arthropathy (SEA syndrome), and gene mutations are pathologically associated. Rheumatology and dermatology share overlapping clinical features. Therefore, it is important to focus on cutaneous manifestations of rheumatological diseases.
We conducted a search at the https://pubmed.ncbi.nlm.nih.gov/advanced/ link with all fields filled in and terms added to the query box. We entered "Dermatological," "Rheumatological," "Children," and "Pediatric" in the "Enter a search term" box. We obtained 48 studies from the years 1980 to 2023. In this review article, we highlight the skin manifestations of common pediatric rheumatological disorders.
2. Evidence Acquisition
2.1. Dermatological Manifestations of Various Rheumatological Disorders
2.1.1. Acute Rheumatic Fever
Acute Rheumatic Fever is caused by group A streptococcal infection (3). Most cases occur in children aged 5 to 15 years. Worldwide, there are approximately 470 000 new cases of ARF. Erythema marginatum is characteristic of ARF but occurs in less than 5% of patients. The mechanism of the skin rash (erythema marginatum) is unknown. Molecular mimicry between streptococcal epitopes and laminin, tropomyosin, vimentin, and keratin plays a role (4). This rash is macular, non-pruritic, non-painful in nature, and may measure up to 0.4 cm in diameter with a serpiginous, erythematous border (5). The most common sites are the trunk, rarely the limbs, and it is exacerbated by warmth (6). The course is intermittent, self-resolving, and does not require any treatment. Subcutaneous nodules are a rare manifestation of ARF and are found in 2% to 10% of patients (Figure 1K). They are more common on bony surfaces like knees, elbows, Achilles tendon, ankles, and the occiput. Their size varies between a few millimeters to 2 cm, and the number varies up to a dozen. They are firm, painless, non-inflamed, and non-adherent to the surrounding tissue (7). They normally subside within 1 - 2 weeks, requiring no treatment (5). Liang et al. found in their mini-review that group B streptococcus can cause a variety of superficial diseases such as pharyngitis, scarlet fever, and impetigo (8).
2.1.2. Sarcoidosis
In childhood sarcoidosis, multiple organs are affected with systemic symptoms such as nausea, fever, weight loss, fatigue, hyporexia, and dysfunction of each affected organ. It is characterized by rash, nodules, ulcers, subcutaneous tumors, and other dermatological manifestations. Subcutaneous nodules, found on extremities, are granulomatous inflammation of adipose tissue underneath the skin (Figure 1J). It occurs in 77% of younger children and 24 - 40% of older children, respectively (9). The rash is erythematous in nature, ranging from red to yellowish-brown or violaceous, with soft, flat-topped papules involving the face, while larger plaques are found on the trunk, buttocks, and extremities. Macular lesions with scarring and ichthyosiform cutaneous manifestations are also seen in children (10).

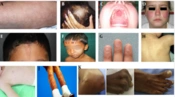
A, Livedo reticularis; B, Scalp with Diffuse Alopecia; C, Oral Ulcer of SLE; D, Malar Rash of SLE; E, En coup de sabre in scleroderma; F, Parry-Romberg syndrome: Atrophy of facial tissues; G, Nail involvement in PsJIA; H, Colored dirty mildly scaly rash in Blau syndrome; I, Salmon pink-macular rash in SoJIA; J, Subcutaneous nodule in sarcoidosis; K, Subcutaneous Nodules in Acute Rheumatic Fever.
2.1.3. Systemic Onset Juvenile Idiopathic Arthritis
Systemic Onset Juvenile Idiopathic Arthritis (SoJIA) (11) accounts for 10% to 30% of children with JIA and is the most common subtype (12). An evanescent, salmon-pink, macular rash (Figure 1I) is found on the trunk and extremities, associated with fever spikes (quotidian fever). Usually asymptomatic, it can be pruritic, with dermographism at sites of scratching or pressure. The immunopathophysiology of SoJIA involves prolonged activation of innate immunity, including neutrophils, monocytes, and macrophages. Proinflammatory cytokines such as IL-1 beta, IL-6, and IL-18 lead to the clinical features of systemic arthritis (13).
2.1.4. Blau Syndrome
Blau Syndrome is characterized by a triad of skin rash, arthritis, and uveitis, and it is inherited as an autosomal dominant disease with a mutation in the NOD2 gene. An erythematous maculo-micropapular fine scaly rash appears on the trunk and extremities, later developing into exanthema which becomes tan-colored and may present a "dirty" scaly appearance (Figure 1H) (14).
2.1.5. Psoriatic Juvenile Idiopathic Arthritis
Psoriatic Juvenile Idiopathic Arthritis (PsJIA) typically begins in fewer than five joints in approximately 80 percent of children, affecting the knee, ankle, and less frequently, the hip joint (15). The most common skin involvement is seen in the form of classic vulgaris followed by guttate psoriasis (16). Psoriasis in children is typically mild, characterized by thin, soft plaques that might appear and disappear, occasionally receding into the intergluteal crease, hairline, umbilicus, or behind the ears (17). Pitting in nails (Figure 1G) is seen in 50 to 80 percent of pediatric cases with Psoriatic JIA, in comparison to 30 percent in childhood psoriasis (18), whereas onycholysis is less common. These nail changes are indicative of enthesitis of the distal insertion of the extensor tendons (19).
2.1.6. Pediatric Scleroderma
Pediatric scleroderma manifests in two forms: Systemic sclerosis (SSc) and localized scleroderma (LS). Systemic sclerosis is characterized by skin, vascular, and visceral organ fibrosis, while LS is characterized by fibrosis of the skin and underlying tissue without deeper internal organ involvement. The former is more common in adults, while the latter affects children. Both disorders share a common pathophysiology of excessive collagen deposition but vary clinically in terms of unique morbidities and prognoses.
2.1.7. Systemic Sclerosis
Raynaud’s phenomenon is a vasospastic phenomenon leading to a triphasic color change: White due to vasoconstriction, blue due to cyanosis, followed by red due to reestablishment of circulation in response to warmth. This leads to swelling, pain, numbness, and tingling sensations. Triggers and structural proteins play key roles in skin sclerosis. Activation of the innate and adaptive immune system by triggers leads to the activation of fibroblast progenitors and lineages. Converted myofibroblasts play a role in the production of extracellular matrix and other protein components associated with sclerosing skin diseases (20).
2.1.8. Localized Scleroderma or Morphea
Localized scleroderma involves the skin, fascia, muscle, and bone. It is classified into various subtypes: Circumscribed morphea (Figure 1E), further divided into two subtypes - superficial and deep; linear scleroderma (Figure 1F), further divided into two subtypes - involving trunk/limbs and head and neck. Affecting the frontal or fronto-parietal region with or without hemifacial atrophy is called en coup de sabre type of morphea. The Parry Romberg syndrome has hemi-atrophy of the face without dermal sclerosis. Other subtypes include generalized morphea, pansclerotic morphea, and mixed morphea. The pathogenesis of LS includes triggers such as viral or bacterial infections, such as by B. Burgdorferi. Genetic factors have also been implicated (21).
2.1.9. Juvenile-onset Systemic Lupus Erythematosus
Juvenile-onset Systemic Lupus Erythematosus (JSLE) is a common systemic autoimmune connective tissue disorder in the pediatric population. Females are four times more affected, with the peak age of presentation being 12 years. The ACR/SLICC criteria include several mucocutaneous manifestations like malar (Figure 1D) and discoid rash, oral ulcers, and photosensitivity. Acute cutaneous lupus erythematosus (ACLE) (22) is characterized by a defined, symmetrical erythematous and edematous, non-pruritic butterfly rash over the nasal bridge, sparing the nasolabial folds. It strongly relates to systemic disease activity in JSLE (22). The term “lupus” describes dermatological conditions that are truly unrelated to lupus erythematosus, often causing confusion for rheumatologists and dermatologists alike (23). Subacute cutaneous lupus erythematosus is rare in JSLE patients (24) but commoner in adults. It includes two forms: Annular/polycyclic lesions and papulo-squamous/psoriasiform lesions, which are characterized as widespread, symmetrical, erythematous papules/plaques with scales and telangiectasia on sun-exposed and non-sun-exposed areas such as the chest and back. The most common sites are the face, upper and lower extremities, and lesions usually heal without scarring (25). Drug-induced lupus cannot be differentiated from normal lupus and is rare in children. Genetic risk factors, environmental exposures, and cellular components of the skin and the innate and adaptive immune systems play a role in the pathophysiology of lupus (26).
Chronic cutaneous lupus erythematosus (CCLE) is uncommon in children below the age of 10 years. Rarely, discoid lupus has been reported in children without systemic symptoms (27). It usually appears as indurated scarring, coin-shaped lesions, purplish papules with atrophic formation, and telangiectasia. These lesions are seen above the neck on the vertex, face, and ears. Photosensitivity may appear as a skin rash reacting to both UVA and UVB light. Diagnosis is mainly based on the patient's history or clinician observations. The lesions occur on sun-exposed areas like the face, upper chest, or extremities.
Oral or nasopharyngeal ulcers represent oral discoid lesions and are nonspecific ulcers (Figure 1C). They begin as solitary erythema and hemorrhagic patches and progress to discoid ulcers with a reticulate border. They are painless and found on the hard palate. They generally represent disease activity (28). Aphthous ulcers are painful, multiple lesions on the buccal mucosa of the oral cavity, lips, and nasal septum. They have a tendency to bleed (29).
Diffuse non-scarring alopecia presents with hair loss without signs of inflammation on the scalp and usually suggests active disease (30). "Lupus hair," which is weak and thin hair around the scalp; patchy non-scarring alopecia, which is moderate erythematous, dispersed patchy hair loss, and alopecia areata, are other types of alopecia observed in JSLE patients (31).
Livedo reticularis (Figure 1A) lesions are commonly seen in juvenile-age patients with anti-phospholipid syndrome (32). It is characterized by discoloration of the skin with a reticulated pattern on the lower extremities.
Raynaud’s Phenomenon: Excessive vasospasms brought on by emotional strain or exposure to cold can cause Raynaud's phenomenon, which is often reversible after rewarming (33).
Bullous SLE: It is rare and most commonly reported in young individuals (34), and is characterized by multiple vesicles and bullae usually on the upper half of the body. Other lesions include calcinosis cutis, acanthosis nigricans, and hypocomplementemic urticarial vasculitis (35).
2.1.10. Juvenile dermatomyositis (JDM)
It is an idiopathic autoimmune inflammatory myopathy characterized by muscular weakness and skin rashes (36). The female-to-male ratio is 5: 1, and the disease typically starts between the ages of 5 and 9 years (36). Gottron papules are thickened, shiny, erythematous plaques commonly found on the dorsal surfaces of the proximal interphalangeal and metacarpophalangeal joints, occasionally appearing on the knees, elbows, and malleoli of the ankle. The heliotrope rash presents as symmetrical erythematous to violaceous coloration on the upper eyelids and may be associated with periorbital edema. Facial erythema is present, sparing the nasolabial folds (Figure 2B). Calcinosis cutis (Figure 2C) refers to dystrophic deposition of hydroxyapatite or calcium phosphate in the soft tissues of the skin, commonly occurring on the buttocks, knees, and elbows. Erythema over the shoulders, upper back, and posterior neck (Figure 2D), known as the shawl sign, and over the upper chest and anterior neck, referred to as the V sign, are also observed. Telangiectasia, indicative of small vessel disease, is commonly visible in nailfolds as tortuous, dilated, or dropout capillary loops, reflecting disease activity (37). The pathogenesis of juvenile idiopathic inflammatory myopathy (JIIM) involves a complex interplay between genetic and environmental factors leading to immunological, vascular, and metabolic dysfunction. Environmental triggers of JIIM include ultraviolet (UV) radiation, pollution, and microbial infections. Genetic loci in the MHC and non-MHC regions contribute to disease susceptibility and development. Type I interferon signaling plays a central role in pathological changes in various tissues, and immune dysregulation within the skin, muscle, and blood vessels is thought to contribute to disease progression (38).

A, Nodular lesion in PAN; B, Skin Rash in JDM; C, Skin calcinosis in JDM; D, Shawl Sign in JDM; E, Urticarial-like appearance in CAPS; F, Acne-like lesions in Behcet's disease; G, Beau lines and induration at BCG inoculation site; H, Rash over extremities, Perianal desquamation, Dry, reddened, and vertically cracked lips; I, Henoch-Schonlein purpura; J, Erysipelas-like erythema, and a full spontaneous in FMF.
2.1.11. Pediatric Vasculitis: Kawasaki Disease (KD)
It is more common in children under five years of age, with a slight male predominance (39). It has an incidence of 60 - 150 per 100 000 children below 5 years of age, making it the most common pediatric vasculitis and the most common vasculitic disorder. The rash is polymorphous, appearing as macular, maculopapular, or morbilliform, starting from the trunk and spreading to the extremities. Desquamation of the rash across the perineal area (Figure 1F) frequently occurs during the first week of illness and usually resolves with the subsidence of fever. Mucosal changes can be seen in the form of dry, reddened, cracked lips, with a characteristic strawberry tongue and erythema of the oral-pharyngeal mucosa. These changes typically occur during the febrile phase of KD. Additional features include the development of erythema in the palms and soles in the acute phase, desquamation at the tips of the fingers and toes in the sub-acute phase, and transverse ridges over the nails known as beau lines (Figure 2G) that may appear in the convalescent phase of KD. The pathophysiology of skin lesions shows edema, dilatation of small vessels in the papillary dermis, and infiltration of CD4+T cells and CD13+ macrophages in the dermis and epidermis. Skin lesions have been shown to contain interleukin-1α and tumor necrosis factor-alpha (TNF-α), which play a role in the pathogenesis of KD (40).
2.1.12. Henoch Schonlein Purpura (HSP)
It is IgA-mediated small vessel vasculitis and is more common in boys. The most common trigger is group-A β-hemolytic streptococci, with other factors including viral infections and drugs. The rash appears over dependent areas such as the ankles and legs, progressing to the buttocks and lower back (Figure 2G). It starts as a pink-reddish maculopapular eruption that becomes palpable and purpuric, fading to a brownish discoloration that persists for weeks. Due to the larger surface area of the head and face in babies and a relatively higher blood supply, lesions are more common in infancy on the face and head (41). The pathophysiology of IgA vasculitis is not fully understood; however, IgA plays a significant role, with immune complexes deposited in the skin causing palpable purpura and petechiae (42).
2.1.13. Polyarteritis Nodosa
It is a medium vessel vasculitis. Classic polyarteritis nodosa (PAN) is uncommon in children, and the etiology of PAN is unknown. It is probably best viewed as an immune complex-mediated disease. It presents with tender subcutaneous nodules, sized between 0.5 - 2 cm, livedo reticularis, and cutaneous ulcerations (43). Patchy, focal livedo reticularis (Figure 2A) in a starburst pattern is considered typical (44). The most commonly affected sites are the legs, arms, and trunk, which may reflect underlying disease, infection, or medication use (45).
2.1.14. Behçet’s Disease
It is a type of variable vessel vasculitis characterized by involvement of small, medium, and large blood vessels. Muco-cutaneous involvement in the form of painful recurrent oral ulcers is common. These ulcers appear yellow-green with a pseudomembranous base (Figure 2F), as round, discrete lesions, and heal without scarring. The most common sites are the tongue, lips, palate, and cheeks. Genital ulcers are less common in children than in adults but are extremely painful and heal with scarring. The scrotum, glans penis, vulva, vagina, and peri-anal region are the most commonly involved sites (46).
2.1.15. Autoimmune Inflammatory Diseases
These are a heterogeneous group of disorders characterized by hyperactivation of the innate immune system independent of an antigen, leading to recurrent or persistent aseptic inflammation. They can present in the form of nonspecific varied cutaneous symptoms. Identification of skin lesions in autoimmune inflammatory diseases (AIDs) may provide clues for diagnosis.
Familial Mediterranean Fever (FMF): Genetically, it is an autosomal recessive autoinflammatory disease, which manifests as frequent fever, abdominal pain, joint pain, swelling, and chest pain. Dermatologic manifestations may be reported in the form of erysipelas-like rashes (Figure 2H) on the lower extremities (leg, ankle, or foot), particularly below the knees, and can be the only presenting symptom (47).
Cryopyrin Associated Autoinflammatory Syndromes (CAPS): It is characterized by pyrexia and inflammation due to dysregulation of innate immunity, such as interleukin 1β. It leads to periodic fever, rash (Figure 2E), musculoskeletal symptoms, seizures, hydrocephalus, conjunctivitis, and pan-uveitis (48).
Pyogenic Arthritis, Pyoderma gangrenosum, and Acne (PAPA) Syndrome: Genetically, it is an autosomal dominant auto-inflammatory condition characterized by repeated, destructive episodes of skin and joint inflammation. Skin manifestations occur in the form of cystic acne, which starts in early puberty. Pyoderma gangrenosum is a severe, life-threatening complication causing large, deep cutaneous ulcerations, mostly on the lower extremities (49).
Synovitis, Acne, Pustulosis, Hyperostosis, Osteitis (SAPHO) Syndrome: It is an uncommon clinical entity, with a combination of osteo-articular and dermatological manifestations. The etiology is thought to be due to mutations in NOD2 and LPIN2 genes present on chromosome 18. Severe acne, hidradenitis suppurativa, palmoplantar pustulosis (PPP), and various types of psoriasis can occur (50).
2.1.16. Infection-Associated Rheumatological Diseases
Lyme disease is a multisystemic disorder spread by the tick, Ixodes, and caused by Borrelia burgdorferi sensu lato, a spirochete. Complications can be prevented by identifying and initiating early treatment, mainly targeting skin lesions. They manifest in the form of erythema chronicum migrans (ECM), borrelial lymphocytoma, and acrodermatitis chronica atrophicans. The primary ECM occurs at the site of a tick bite and develops within 1 - 3 weeks. It exists in two forms: (1) Enlarges with various hues of erythema; (2) centrifugal spread with central clearing and a bulls-eye appearance, with a bite mark at the center, giving it a targetoid appearance. Lesions typically resolve spontaneously within weeks to months, though burning, pruritus, or pain can occasionally occur. Acrodermatitis chronica atrophicans is a late cutaneous manifestation of Lyme disease, often affecting acral skin due to cooler temperatures, mainly on the extensor surfaces and bony prominences. Healing of these lesions does not occur spontaneously. Borrelia Lymphocytoma, the least common manifestation, may develop after 30 - 35 days or sometimes even months later following a bite. Poorly defined bluish-red patches develop, which later spread and convert into well-defined plaques, typically appearing on the earlobe in infants (51).
2.1.17. Paradoxical Skin Reactions
A mini-review by Garcovich et al. revealed an unexpected occurrence of paradoxical inflammation during the use of biological agents, which has become a new type of drug-related adverse event. These reactions affect the skin and present a spectrum of clinical aspects (51).
The common skin conditions and dermatological manifestations can be differentiated by the age of presentation, disease course, progression, associated clinical features, family history, and investigations. It is a combination of all these factors but not a single clinical or lab parameter that helps in differentiation.
The strength of our study is that it contributes to the understanding of this particular topic. Limitations include its lack of originality and reliance on existing studies that may not be exactly similar. Further original studies are required to evaluate the dermatological signs associated with rheumatological illnesses in children.
3. Conclusions
Rheumatic diseases may present with dermatological manifestations in children. Detailed history and careful examination of such cases by pediatric rheumatologists are essential for accurate diagnosis and early treatment of the diseases. A combined approach involving pediatric rheumatologists and dermatologists may be helpful, and further original studies are required to establish the role of this combined approach (Table 1).
| No | Name of Disease | Epidemiology | Pathophysiology | Clinical Presentation | Dermatological Manifestations |
|---|---|---|---|---|---|
| 1 | Acute Rheumatic Fever | Caused by group A streptococcal infection. Common age is 5 to 15 years, there are 470 000 new cases of ARF annualy | Largely unknown. molecular mimicry between streptococcal epitopes and laminin, tropomyosin, and others may play a role | Pyoderma, Fever, tonsillopharyngitis arthritis, arthralgia, carditis, erythema marginatum, Rash, skin nodules, sydnehems chorea | Pyoderma, erythema marginatum, Rash, skin nodules |
| 2 | Sarcoidosis | Mainly in (77%) the young children and 24 - 40% in older children | Granulomatous inflammation of adipose tissue | Multiple organs are affected with clinical features s such as fever, weight loss, fatigue, hyporexia, and nausea | Rash, nodules, ulcers, subcutaneous tumours |
| 3 | Henoch Schonlein Purpura | Common vasculitis in children, affecting 10 to 20 children per 100 000 annually male-to-female ratio of 1.5:1, and commonly affects children aged 3 to 8 years. | Not fully understood; however, IgA and Immune complexes deposited in the skin may have role | Abdominal pain arthritis or arthralgia, proteinuria, hematuria, and leukocytoclastic vasculitis other common symptoms are fever, scrotal pain, and edema in boys, and rarely | Rash appears over ankles and legs progressing to buttocks and lower back. Papular palpable purpuric eruption fading as a brownish discoloration |
| 4 | Juvenile dermatomyositis (JDM) | Most coomon age of onset is 4 and 10 years. More common in girls than boys (2: 1). incidence of JDM is 3 per million children per year, | A, ultraviolet radiation, pollution and infections. B, Genetic MHC and non-MHC loci C, Type I interferon D, Immune dysregulation in the dermal, muscular and vascular systems. | Bilateral proximal muscle weakness. Weakness is insidious at onset. Arthralgia or arthritis and Rash | Heliotrope rash, Gottron’s papules, periungual erythema. Facial erythema crossing the nasolabial folds is common shawl sign, V sign and calcinosis |
| 5 | Polyarteritis nodosa (PAN) | Exact incidence, and prevalence, of CPAN are not known. A study from reported incidence 0.7 per million children. | Probably as an immune complex-mediated disease due to underlying disease, infection, or medication | Subcutaneous nodule, livedo reticularis and cutaneous ulcerations. Common sites are legs, arms and trunk. | Tender subcutaneous nodule, , livedo reticularis and cutaneous ulceration. Patchy, |
| 6 | Systemic sclerosis | Incidence ranging from 0.45 to 1.9 per 100 000, mean age ofonset is 8.1 years, children under 16 years of age account only than 5% of total cases, fourfold more frequent in females | Both immune system by e triggers is lead to activation of fibroblast progenitors and lineages. Excessive amounts of matrix and other protein components lead to sclerosing skin diseases | There is sl thickening and hardening of the skin with changes in internal organs, Arthralgia and arthritis | Sclerosis/induration proximal to metacarpophalangeal joint, Raynauds phenomenon, skin rash |
| 7 | Localised sclerodema | Autoimmunity, HLA class I and II alleles, environmental triggers, infections, and other factors. | Triggered by viral or bacterial infection, such as by B. Burgdorferi. Genetic factors | Predominant scleroderma which affects the skin. underlying fascia, muscle, joints and bone may also affected. | Linear indurated plaques with dyspigmentation on the limbs, on head it is known as en coup de sabre, |
| 8 | SLE | Chronic autoimmune disease | Genetic factors, environmental exposures, and cellular components and innate and adaptive immune systems | Multisystem progressive organ involvement mainly musculoskeletal, skin, kidney, hematologic, neurological and Psychiatric and development of antinuclear and other antibodies | Hall-mark of malar, or butterfly rash. It is erythematous, raised, non-pruritic, it extends over the nasal bridge, to chin and ears, usually it spares the nasolabial folds |
| 9 | Kawasaki Disease | Affects children under 5 years, incidence of KD was 19.1 per 100 000 among children less than 5 years old | Small vessels dilatation and edema, invasination of CD4+T, CD13+ cells in dermis and epidermis interleukin-1α and tumor necrosis factor alpha (TNF-α) | Fever, skin rash in extremities, Polymorphous exanthema, conjuctival injection in both eyes, lips and oral cavity color and cahnges, Cervical lymphadenopathy | Maculopapular erythematous rash with a ‘perineal accentuation’. Scarlatiniform rash, erythema multiforme-like rash with target lesions. |
| 10 | Systemic Onset Juvenile Idiopathic Arthritis | Incidence is 1.6 - 23 cases for 100 000 children, relative risk of JIA in siblings varies from 15 to 30 | Prolonged activation of innate immunity, innate proinflammatory cytokines including IL-1 beta, IL-6, and IL-18 leads to the disease | Joint pain, arthritis, fever, Rash, Lymphedenopathy, hepato-splenomegaly, Serositis, seizures, meningismus, dearanged lab parameters | Evanescent rash associated with fever spikes. It can be pruritic, with dermographism at sites of scratching |
Informative Details Are Summarized from
