1. Introduction
Wells syndrome (WS), also known as eosinophilic cellulitis, was initially termed "recurrent granulomatous dermatitis with eosinophilia" by Wells over 40 years ago (1). The nomenclature was later simplified to "eosinophilic cellulitis" by Wells and Smith, a term commonly used today (2). This inflammatory dermatosis, recognized for its rarity, exhibits a wide clinical and histological spectrum, with its precise etiopathogenesis remaining uncertain (3), despite documented links with various infectious diseases.
We describe the case of a young woman suffering from generalized Wells syndrome with various lesion types linked to an uncommon infectious cause. This emphasizes the importance of dermatologists recognizing and diagnosing this disease early to avoid erroneous or delayed diagnosis and its consequences, such as worsening symptoms, sleep disturbance, and poor quality of life.
2. Case Presentation
A 39-year-old female patient with a past medical history of obesity and hypertension presented with a one-month history of erythematous lesions with desquamation, initially appearing in the armpits and inframammary fold, and subsequently spreading to the abdomen, neck, and antecubital fold, accompanied by mild pruritus. The patient had used nutratopic neutral soap and nutratopic moisturizing cream, hypoallergenic laundry soap, betamethasone cream, and clobetasol cream twice daily for a week, as well as oral prednisolone once daily for 5 days, resulting in partial improvement.
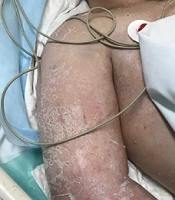
Upon physical examination, the patient, with Fitzpatrick III phototype, had erythematosquamous plaques with well-defined irregular borders on the face, neck, armpit crease, and inframammary region. Additionally, erythematous plaques with whitish desquamation were noted on the abdomen, while annular erythematous plaques with fine whitish desquamation inside were present on the thighs (Figure 1).

Figure 1.
Clinical presentation A - B, erythematous plaques with desquamation, characterized by well-defined irregular borders on the neck and armpits; C, notably, on the abdomen, multiple small erythematous plaques with whitish desquamation are present along the inner edge; D, furthermore, on the thighs, one can observe multiple annular erythematous-squamous plaques.
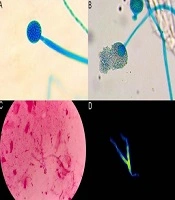
Initially, the diagnostic impression included typical pityriasis rosea with distinctive morphology in folds or conditions like tinea corporis or allergic contact dermatitis in folds. Laboratory tests indicated normal blood counts, except for hypereosinophilia. Tests for syphilis and antinuclear antibodies were negative, and armpit and trunk KOH tests were also negative. However, a positive mycological culture for skin was obtained for Rhodotorula glutinis/Rhodotorula mucilaginosa.
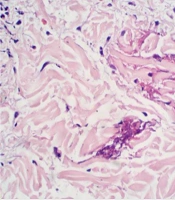
A skin biopsy and subsequent histopathological examination were performed to determine the etiology. The findings revealed superficial and medium dermatitis, predominantly characterized by superficial cells composed of lymphocytes accompanied by numerous eosinophils in a perivascular and focally interstitial location. Additionally, slight edema was noted in the superficial dermis, along with spongiotic epidermal changes mainly affecting the lower half of the epithelium. The examination also indicated exocytosis of some eosinophils, laminar hyperkeratosis, and parakeratosis in mounds. Moreover, discrete vacuolar changes in the interface, extravasated red blood cells, and fibrinoid collagen necrosis resembling flame-like figures were observed (Figure 2).

Figure 2.
The hematoxylin-eosin staining displayed flame figures in the superficial dermis, indicative of the underlying pathology.
Upon careful evaluation of the clinical and histopathological findings, the diagnosis of Wells syndrome was conclusively established.
3. Discussion
Wells syndrome is a dermatosis that can manifest at any age, from infancy to old age, with a higher incidence noted among young adults. There is no distinct gender predilection, as 55% of cases occur in women and 45% in men (2). The exact etiology remains unknown; however, it is believed to involve a type IV hypersensitivity reaction triggered by various factors, leading to eosinophil chemotaxis and degranulation (2).
The triggers for this reaction include infections (bacterial, fungal, and parasitic), insect bites, asthma, vaccines, drugs (such as antibiotics, anticholinergics, anesthetics, and non-steroidal anti-inflammatory drugs), autoimmune diseases (like ulcerative colitis), hematological disorders (e.g., chronic lymphocytic leukemia, Hodgkin's disease), surgical procedures, and solid tumors (such as nasopharyngeal and renal cell carcinoma) (1-4).
In our patient, we considered an uncommon cause: A fungal infection for which only two documented instances exist in the literature (5, 6). The precise mechanism by which an immune response resembling that of a fungal infection induces dermatitis with eosinophilic infiltration, a hallmark of Wells syndrome, remains unclear, as does the significant clinical variability associated with it.
The clinical presentation of Wells syndrome is highly diverse, including infiltrated erythematous plaques, urticaria or erysipelas/cellulitis-like lesions, and granulomatous or morphea-like lesions (1). Some cases have been reported with papules, plaques, vesicles, or blisters resembling granuloma annulare (1, 7). Symptoms may include a burning sensation, pruritus, or even systemic symptoms such as fever, arthralgia, general malaise, tissue (in 61% of cases), and blood hypereosinophilia, as well as increased immunoglobulin E and erythrocyte sedimentation rate (1-3).
The involvement can be localized, occurring in 59% of cases, which is more common in children, or generalized, occurring in the remaining 41% of cases, which is more common in adults and is related to a more severe and prolonged clinical course (2).
Hattori and Seishima reported a case of a young woman who had recurrent erythema of the left thigh for 2 years. She then developed papules and plaques with scaling, some of which had exudate, throughout her body. The patient had hypereosinophilia, and a biopsy showed dense cellular infiltration of mononuclear cells and eosinophils throughout the dermis and subcutaneous tissue, with flame figures. Her culture also revealed Trichophyton rubrum (6). Similarly, Zanini in Brazil reported a young patient with multiple excoriated violaceous papules, ulcerations, some vesicles, pustules, and erythematous nodules on the buttocks, with a direct mycological examination positive for septate hyphae. The patient was treated for tinea corporis incognita with improvement, but 2 months later, the lesions recurred. A skin biopsy showed intense eosinophilic infiltrate with the presence of flame figures (5). These cases demonstrate the wide range of clinical findings associated with Wells syndrome.
We found these two case reports of Wells syndrome associated with fungal infection, similar to our case. In all cases, the patients were young, healthy individuals with multiple skin lesions, such as generalized scaly plaques and vesicles, and the conditions were recurrent. The histopathology in each case reported distinctive characteristics of Wells syndrome. Regarding differences, the fungi reported in cultures were different: Trichophyton in the previous cases and Rhodotorula glutinis in our case. Additionally, the treatments varied, with triamcinolone infiltration and topical antifungal management in the reported cases, while our patient was treated with topical corticosteroids, antihistamines, and oral antifungal treatment.
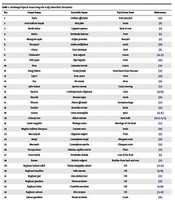
Wells syndrome is considered a diagnosis of exclusion. Therefore, a comprehensive medical history, blood tests to rule out systemic diseases, and histopathological confirmation are necessary to differentiate it from conditions like urticaria, cellulitis, contact dermatitis, fixed drug rash, granuloma annulare, erythema elevatum diutinum, and Churg-Strauss syndrome, among others (1, 8, 9). While diagnostic criteria have been proposed, they require validation in larger patient cohorts (1) (Table 1). When applying these criteria to our patient, they were found to be met.
| Major Criteria | Minor Criteria |
|---|---|
| Typical clinical features | Histology with flame figures |
| Chronic relapsing course | Histology with granulomatous changes |
| Exclusion of systemic disease | Peripheral eosinophilia up to 1500/uL |
| Histology: Eosinophilic infiltrates, no signs of vasculitis | Detectable triggering factor |
a Requires 2 major and 1 minor criteria.
In histopathology, the results exhibit variability depending on the timing of the biopsy, leading to the identification of three distinct phases (2). Initially, there is an acute phase characterized by edema and extensive dermal infiltration of eosinophils. Subsequently, during the subacute phase, eosinophils degranulate in the vicinity of collagen bundles, resulting in the distinctive "flame shape" appearance frequently observed in documented cases. Finally, in the resolution phase, histiocytes and giant cells replace eosinophils surrounding the remaining flame figures, giving rise to granulomas (2, 3).
It is important to acknowledge that while flame figures are indicative of Wells syndrome, they are not pathognomonic (4). These figures can manifest in various other conditions, including pemphigoid, pemphigus, Churg-Strauss syndrome, gestational herpes, eczema, drug rash, among others. Consequently, establishing a clinical-pathological correlation is essential in arriving at a definitive diagnosis (4).
Most cases of Wells syndrome resolve spontaneously. However, if a trigger is found, etiological treatment can be effective (8). Systemic and/or topical corticosteroids are the first-line and most efficient treatment for Wells syndrome (3, 8). The use of antihistamines is ideal for the adjuvant management of pruritus (2).
For patients demonstrating resistance to corticosteroids or facing intolerable adverse effects from prolonged therapies, alternative treatment modalities include dapsone, tacrolimus, ciclosporin, minocycline, tetracycline, griseofulvin, methotrexate, and azathioprine (3, 10, 11). Recent reports have highlighted the use of tumor necrosis factor (TNF)-α inhibitors, such as adalimumab. Although its mechanism of action is not well understood, it may play a role in eosinophil recruitment and IgE production (2, 3). In our patient's case, treatment with topical corticosteroids, antihistamines, and itraconazole initially showed a good response; however, lesions appeared in other sites, leading to the adjustment of treatment with systemic corticosteroids, which resulted in resolution.
The overall prognosis of the disease is favorable, with most patients experiencing resolution of skin lesions within weeks to months. However, some patients may have a poor prognosis if they have recurrent lesions without a clear etiology or associated diseases such as hematological neoplasms, Churg-Strauss syndrome, or hypereosinophilic syndrome (3).
5.1. Conclusions
Wells syndrome is a rare skin disorder with polymorphic manifestations, such as papules, plaques, vesicles, or blisters, making diagnosis challenging in clinical practice. Therefore, a high index of suspicion is required, along with blood tests and skin biopsy, to confirm the diagnosis and identify the etiology for a more specific treatment approach. This will help avoid a late or erroneous diagnosis. The prognosis of the disease is generally favorable because the lesions resolve spontaneously in the majority of patients.