



1. Context
Pathologically, it can lead to cardiac myocyte death due to chronic ischemia. Reports indicate that coronary artery disease (CAD) causes approximately seven million deaths worldwide each year (1). Myocardial infarction (MI) imposes a substantial economic burden. Hospitalization costs alone in the United States amount to at least $450 billion, and patients lose many years of healthy life (2).
To preserve myocardial health, it is crucial to restore blood flow quickly during ischemia (3). Reperfusion interventions such as primary percutaneous coronary intervention (PCI) and thrombolysis have been shown to reduce mortality and infarct size and improve left ventricular function. However, reperfusion can also lead to complications (4). Swift restoration of coronary flow can result in reversible myocardial contractility (myocardial stunning), ventricular arrhythmias, and microvascular dysfunction.
Myocardial ischemia/reperfusion (I/R) injury follows a pattern of damage, with the adverse effects of cardiac ischemia/reperfusion leading to myocyte necrosis and heart failure (5).
One of the yet-to-be-discovered molecular mechanisms of IR damage in muscles is the production of reactive oxygen species (ROS). Excessive ROS accumulation activates the mitochondrial permeability transition pore, causing cellular oxidative stress, mitochondrial dysfunction, and cell death. Elevated ROS levels may reduce myofibrillar Ca2+ sensitivity, impairing muscle contractile function. Additionally, modest ROS levels can act as signaling molecules in protective ischemia preconditioning (IPC) pathways (6).
Antioxidant treatment has shown variable and inconsistent effects in experimental and clinical studies, highlighting the need for further research in this field. Cohort studies, however, consistently show a connection between antioxidants such as flavonoids and beta-carotene and reduced cardiovascular disease (CVD)-related mortality (1).
2. Evidence Acquisition
Novel cardioprotective strategies for enhancing myocardial salvage and cardiac function are of paramount importance. Recently, plant-based medicines with antioxidant properties, such as grape seed extract and its derivatives like epigallocatechin or Gallic acid, have gained attention (7). Consequently, we have embarked on investigating their molecular mechanisms as a therapeutic alternative to conventional pharmacological approaches, which often come with numerous side effects (8).
3. Results
3.1. Inorganic Nitrate or Nitrite Therapy
Inorganic nitrate or nitrite therapy has shown significant reductions in oxidative stress, heart hypertrophy, fibrosis, and blood pressure levels while also improving renal function. In unilaterally nephrectomized rats, treatment with a high dose of sodium nitrate (140 - 1400 mg/kg/day) resulted in decreased mean arterial pressure, cardiac hypertrophy/fibrosis, plasma malondialdehyde, isoprostane, and 8-oxo-2-deoxyguanosine levels, along with improved kidney morphology (9).
Using a lower dosage of sodium nitrite (15 mg/kg/day), spontaneously hypertensive rats exhibited lower blood pressure and improved endothelial function. These blood vessels produced more nitric oxide (NO), leading to increased cGMP production and enhanced endothelial nitric oxide synthase (eNOS) activity (10). Sodium nitrite at a dose of 7.5 mg/kg/day significantly lowered systolic blood pressure and reduced oxidative stress in the vasculature and kidneys in angiotensin-II-infused mice, a model of arterial hypertension (11).
The same study demonstrated that sodium nitrite (15 mg/kg/day) diminished systolic blood pressure, malondialdehyde levels, and vascular dihydroethidium in rats treated with deoxycorticosterone (DOCA) and salt (12).
Using inorganic nitrite/nitrates has been found effective in reducing inflammation and inhibiting oxidation, including the upregulation of arginase and pathways dependent on cyclic guanosine monophosphate (cGMP)/cyclic adenosine monophosphate (cAMP) (13). For example, supplementation with inorganic nitrite/nitrate prevented microvascular inflammation in rats and mice treated with non-steroidal anti-inflammatory drugs (NSAIDs), leading to a reduction in tumor necrosis factor-alpha (TNF-α) expression and leukocyte recruitment (14). In rats and mice, inorganic nitrite/nitrate supplementation reduced the production of tumor necrosis factor-alpha (TNF-α) and adhesion molecules, as well as leukocyte recruitment, which was enhanced by NSAID treatment (15). In lipopolysaccharide (LPS)-stimulated phagocytes (macrophages), nitrite directly inhibited NADPH oxidase, mediated by xanthine oxidoreductase-dependent bioactivation to nitric oxide (NO), leading to the downregulation of protein-level cytosolic NADPH oxidase subunits p47phox and p67phox but not mRNA (16).
In Apo E knockout animals fed a high-fat diet, inorganic nitrate reduced leukocyte rolling, recruitment to the vascular wall, and myeloperoxidase activity (17). In active neutrophils, inorganic nitrite inhibited the production of proxy nitrite and hypochlorite. However, the specific mechanism by which inorganic nitrite and nitrate inhibit immune cell activation remains unknown (18).
3.2. Cardioprotective cGMP–PKG Pathway
The cGMP–PKG pathway is initiated by nitric oxide (NO) and natriuretic peptides (NPs). Nitric oxide initiates this signaling pathway by activating soluble guanylate cyclase (sGC), a heterodimeric enzyme composed of α- and β-subunits with prosthetic heme groups. Atrial NP (ANP), brain NP (BNP), and C-type NP (CNP) activate particulate GC (pGC) in the plasma membrane to produce cGMP.
The second messenger, cGMP, is generated by both sGC and pGC. However, the downstream functions of cGMP vary depending on its subcellular location. High intracellular levels of cGMP primarily mediate physiological actions through protein kinase G (PKG), which is cGMP-dependent. PKG-I, the original kinase in animals, plays a crucial role in transmitting physiological signals to the cardiovascular system. Notably, PKG-I and PKG-1 differ in their N-termini due to alternative splicing. Research indicates that PKG-I is ten times more sensitive to cGMP than PKG-1. Additionally, PKG-I can be activated independently of cGMP through thiol oxidation, thanks to its unique cysteine residue (Cys 42). This cGMP-independent activation of PKG-1 can reduce blood pressure by promoting vasodilation, though its effects on cardiomyocytes are not fully understood.
Numerous studies have identified multiple downstream effectors of cGMP-PKG-I in the cardiovascular system. For instance, in cardiomyocytes, PKG-I phosphorylates phospholamban at Ser16, activating the sarcoplasmic reticulum Ca2+-ATPase (SERCA). This activation enhances Ca2+ uptake into the sarcoplasmic reticulum during systole, reducing cytosolic Ca2+ levels. Additionally, cGMP-induced PKG-I activation has been linked to the opening of mitochondrial ATP-sensitive K+ (mitoKATP) channels in the inner mitochondrial membrane, leading to an increase in K+ influx and mitochondrial matrix alkalinization. This process boosts H2O2 production from complex I and subsequently activates protein kinase C (PKC), protecting cardiomyocytes against cell death by inhibiting the opening of mitochondrial permeability transition pores (MPTP).
Furthermore, the cardiac myocyte Ca2+-Activated BK type K+ channels (CMBK) play a crucial role in cardiac remodeling following myocardial infarction (MI). CMBK deficiency has been associated with more severe myocardial injury after ischemia/reperfusion (I/R), characterized by increased reactive oxygen species (ROS) production. In addition, angiotensin II (ATII)-induced cardiac hypertrophy, mediated by ROS and mitogen-activated protein kinase (MAPK) activation, was found to be mitigated by antioxidant treatment.
Changes in ion flow, mediated by ROS, have also been linked to the regulation of muscle contractility in cardiomyocytes. Increased ROS production reduces myofibrillar calcium (Ca2+) sensitivity and inhibits sarcoplasmic reticulum Ca2+ ATPase 2 activity (SERCA2), potentially disrupting excitation-contraction coupling and accelerating heart failure progression.
While the detailed mechanistic interactions between downstream effectors and the cGMP-PKG pathway in maintaining heart health require further characterization, numerous preclinical studies have demonstrated the cardio-protective potential of this pathway through pharmacological interventions and gene manipulations. Based on the available evidence, strategies involving either enhanced cGMP synthesis (e.g., sGC activators or stimulators) or reduced cGMP degradation (e.g., PDE inhibitors) represent promising therapeutic approaches for targeting this system.
3.3. Ginsenoside Rb1
Ginsenoside Rb1 (G-Rb1) is a component of ginseng, an herbal remedy with a centuries-old history of use worldwide, particularly in Asian countries. Ginseng contains various compounds known as ginsenosides, which are triterpene saponins. To date, researchers have identified over 30 distinct types of ginsenosides.
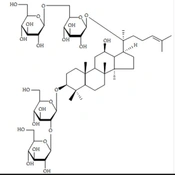
Ginsenoside Rb1 (G-Rb1) exhibits numerous beneficial effects within the cardiovascular system, likely attributable to its antioxidant, anti-inflammatory, and anti-apoptotic properties. These attributes enhance its angiogenesis-promoting and antiarrhythmic effects. In cases of acute myocardial infarction (MI), G-Rb1 can prevent ventricular hypertrophy, as illustrated in Figure 1.

Chemical structures of ginsenoside Rb1
The following are potential mechanisms by which G-Rb1 may mitigate myocardial ischemia/reperfusion (I/R) injury: (1) Inhibition of apoptosis: G-Rb1 achieves this by down-regulating the expression of caspase-3 (19); (2) inhibition of the p38MAPK signaling pathway (20); (3) enhancement of antioxidant defenses: G-Rb1 elevates antioxidant levels, including glutathione (GSH) and superoxide dismutase (SOD), reducing malondialdehyde (MDA) levels-a marker of oxidative stress. This process is likely linked to chondrogenesis attenuation (21); (4) suppression of inflammation: G-Rb1 inhibits the expression of tumor necrosis factor-alpha (TNF-α) by preventing the activation of the PI3K/Akt signaling pathway and interfering with p38MAPK signaling. This crosstalk between the two pathways contributes to its anti-inflammatory effects (21); (5) promotion of angiogenesis: G-Rb1 increases the expression of vascular endothelial growth factor (VEGF) and enhances circulation by elevating nitric oxide (NO) levels through the activation of the PI3K/Akt signaling pathway (22, 23).
3.4. Redox Mechanisms of IPC in Cardiac Muscle
Ischemic preconditioning (IPC) has been demonstrated to protect both cardiac and skeletal muscles from ischemia-reperfusion (IR) injury by subjecting them to sub-lethal levels of IR for several cycles (24). However, the exact mechanism behind IPC remains unknown despite it being one of the most effective methods for protecting against ischemic injury in both experimental and partially clinical studies (25).
During prolonged exposure to IR, preconditioned myocardium exhibits reduced oxidative stress (26). The release of ATP-sensitive K+ (mitoKATP) channels and the limited production of reactive oxygen species (ROS) play a role in IPC's protective effect. Disruption of either mitoKATP channels or ROS production can counteract IPC's safety net (27). Activation of mitoKATP channels may result in the generation of mitochondrial ROS, which prevents cell death by inhibiting mitochondrial permeability transition pore (mPTP) opening through protein kinase C (PKC) activation (28). Protein kinase C serves as a signaling molecule that protects mitochondria throughout the IPC process. In the early stages of IPC, activation of opioid, bradykinin, and adenosine receptors leads to PKC activation and sensitizes the A2b adenosine receptor (A2bAR). This sensitization triggers a survival pathway involving PI3 kinase, Akt, and ERK, ultimately blocking mPTP opening (29). MitoKATP activation has also been observed to inhibit mitochondrial matrix contraction, potentially enhancing ATP production (6).
3.5. Molecular Mechanisms of H2S-Induced Cardio Protection
With advances in scientific technology, researchers have uncovered the role of hydrogen sulfide (H2S) in various physiological and pathological processes in mammals. H2S, akin to nitric oxide (NO) and carbon monoxide (CO), is now recognized as a novel gaseous signaling molecule (30). H2S is endogenously produced by several enzymes, including cystathionine-synthase (CBS), cystathionine-lyase (CSE), 3-mercaptopyruvate sulfur-transferase (3-MST), and cysteine aminotransferase (CAT) (31). The expression of these enzymes varies depending on the tissue. CBS is a crucial enzyme for H2S production in the nervous system, while CSE is the primary H2S-producing enzyme in the cardiovascular system (32).
Numerous studies have shown that H2S is associated with various pathophysiological processes such as oxidative stress, inflammation, apoptosis, and angiogenesis (30). Increasing evidence suggests that H2S plays a significant role in regulating cardiac functions and has a protective effect on the pathophysiology and development of heart disorders.
In this context, we will elucidate the mechanisms underlying this protective role, particularly in the context of preventing arrhythmias, which are undesirable side effects of ischemia that H2S has been shown to guard against.
3.6. Regulation of Ion Channels
Hydrogen sulfide (H2S) has significant effects on heart electrophysiology. The myocardial membrane contains two different types of Ca2+ channels, namely L-type and T-type. L-type Ca2+ channels are essential for maintaining the electrophysiological basis for the plateau phase of action potentials and excitation-contraction (EC) coupling (33). Whole patch clamp experiments in rat cardiomyocytes have revealed that NaHS negatively regulates L-type Ca2+ channels composed of CaV1.2 subunits (34).
In various clinical conditions such as cardiac hypertrophy and heart failure, T-type Ca2+ channels can be observed in atrial and ventricular myocytes, contributing to aberrant electrical activity and EC coupling (35). Recent research has confirmed that NaHS selectively inhibits T-type Ca2+ channels heterologously expressed in HEK293 cells (10 μM - 1 mM) (36).
KATP channels are distributed on the cell membranes and mitochondria in the myocardium and play a crucial role in the principal endogenous cardioprotective mechanism involved in cardiac ischemia preconditioning-opening of KATP channels. Opening of KATP channels leads to hyperpolarization, reducing calcium influx via L-type Ca2+ channels and preventing Ca2+ overload. Tang and coworkers have reported that NaHS (100 μM) opens KATP channels in vascular smooth muscle cells (37). Additionally, it may indirectly activate KATP channels by causing intracellular acidosis (38). H2S shortens action potential duration (APD) and exerts cardioprotective effects by activating KATP channels, while it does not affect the amplitude of action potentials or resting potential (39).
Studies have indicated that H2S can modulate voltage-dependent Na+ channels (Nav). There are native Nav and recombinant Nav (NaV1.5) expressed in different tissues, including the heart. Strega et al. demonstrated that NaHS altered peak sodium current, voltage dependence of inactivation, and activation in Nav channels (40). Since NaV1.5 is responsible for the upstroke of the cardiac action potential, H2S might have a similar impact on NaV channels expressed in the heart.
Chloride channels play a significant role in the normal physiological functions of myocardial cells. However, abnormal changes may occur in pathological conditions such as myocardial ischemia and arrhythmias. Malenkov et al. proposed that H2S affects single-channel currents of chloride channels, showing that NaHS inhibits chloride channels by reducing channel open probability in a concentration-dependent manner, as demonstrated by patch clamp experiments (41). The biological effects of H2S in the heart may be linked to its inhibitory impact on chloride channels (42).
3.7. Pyruvate's Cardio-Protective Antioxidant Mechanisms
In addition to its ATP-producing activities, pyruvate exhibits potent antioxidant effects as a metabolic fuel. The antioxidant properties of pyruvate are mediated through various pathways, as illustrated in Figure 2 (43). Through direct, non-enzymatic processes, pyruvate-containing aliphatic carboxylates undergo oxidative decarboxylation, effectively reducing and detoxifying peroxides, yielding electrons, hydroxyl radicals, and nitrite substitutes (44). Consequently, pyruvate has several effects: (1) Reduction of lipid peroxides to their conjugated alcohols, thereby preventing membrane lipid peroxidation; (2) conversion of hydrogen peroxide (H2O2) and hydroxyl radicals (OH) into water; and (3) transformation of peroxynitrite into a benign form of nitrite (45).
Pyruvate decarboxylation also generates acetate, an oxidizable fuel. Glutathione, a tripeptide and the most abundant natural antioxidant in cells contains a sulfhydryl moiety capable of neutralizing peroxides, peroxynitrites, and hydroxyl radicals (46). During ischemia, particularly during efforts to restore circulation, such as revascularization and fluid resuscitation, large quantities of these hazardous metabolites are generated (47). The oxyradical load can deplete cellular glutathione (GSH), jeopardizing GSH-dependent antioxidant mechanisms. Pyruvate supports GSH in two ways, as depicted in Figure 2 (48).
 Pyruvate acts non-enzymatically to detoxify peroxynitrite (ONOO<sup>-</sup>) into nitrite and peroxides (ROOH) into their corresponding alcohols (ROH); (2) Through the lactate dehydrogenase (LDH) equilibrium, pyruvate oxidizes cytosolic NADH, replenishing NAD<sup>+</sup> for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and reducing the formation of superoxide (. O<sup>2-</sup>) by NAD(P)H oxidase (NOX); (3) Pyruvate undergoes carboxylation by malic enzyme, leading to an increased concentration of Krebs cycle intermediates, such as citrate and isocitrate, within the mitochondria. This supports the production of NADPH by mitochondrial NADP-dependent isocitrate dehydrogenase (ICDH); (4) Citrate is transported to the cytosol via tricarboxylate transporters (TCT) and converted into isocitrate by aconitase (ACON), providing a substrate for NADPH generation by cytosolic ICDH. (5) Citrate inhibits phosphofructokinase (PFK), diverting glycolytic flux into the pentose phosphate pathway (PPP). (6) NADPH, produced by mechanisms 3, 4, and 5, serves as a reducing agent to convert glutathione disulfide (GSSG) back into glutathione (GSH). This helps maintain the redox state of GSH for the activity of glutathione peroxidases (GPX) and other GSH-dependent antioxidant systems (Abbreviations: CS, citrate synthase; DAP, dihydroxyacetone phosphate; DPG, 1,3 bis-phosphoglycerate; GAP, Glyceraldehyde 3-phosphate; GR, glutathione reductase; MCT, monocarboxylate transporter; PDH, pyruvate dehydrogenase; PGI, phosphor glucose isomerase. Please refer to the main text for further details).")
The antioxidant mechanisms involving pyruvate: (1) Pyruvate acts non-enzymatically to detoxify peroxynitrite (ONOO-) into nitrite and peroxides (ROOH) into their corresponding alcohols (ROH); (2) Through the lactate dehydrogenase (LDH) equilibrium, pyruvate oxidizes cytosolic NADH, replenishing NAD+ for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and reducing the formation of superoxide (. O2-) by NAD(P)H oxidase (NOX); (3) Pyruvate undergoes carboxylation by malic enzyme, leading to an increased concentration of Krebs cycle intermediates, such as citrate and isocitrate, within the mitochondria. This supports the production of NADPH by mitochondrial NADP-dependent isocitrate dehydrogenase (ICDH); (4) Citrate is transported to the cytosol via tricarboxylate transporters (TCT) and converted into isocitrate by aconitase (ACON), providing a substrate for NADPH generation by cytosolic ICDH. (5) Citrate inhibits phosphofructokinase (PFK), diverting glycolytic flux into the pentose phosphate pathway (PPP). (6) NADPH, produced by mechanisms 3, 4, and 5, serves as a reducing agent to convert glutathione disulfide (GSSG) back into glutathione (GSH). This helps maintain the redox state of GSH for the activity of glutathione peroxidases (GPX) and other GSH-dependent antioxidant systems (Abbreviations: CS, citrate synthase; DAP, dihydroxyacetone phosphate; DPG, 1,3 bis-phosphoglycerate; GAP, Glyceraldehyde 3-phosphate; GR, glutathione reductase; MCT, monocarboxylate transporter; PDH, pyruvate dehydrogenase; PGI, phosphor glucose isomerase. Please refer to the main text for further details).
Pyruvate safeguards cellular GSH levels through two primary mechanisms: First, by directly detoxifying RONS, it unburdens GSH-dependent antioxidant enzymes. Second, carboxylation of pyruvate increases the amount of Krebs cycle intermediates, such as citrate, which is a natural inhibitor of the glycolytic enzyme phosphofructokinase (49).
In isolated hearts and in vivo myocardium, pyruvate treatment significantly elevates citrate content (50). Subsequently, citrate is transported into the cytosol by tricarboxylate transporters in the inner mitochondrial membrane (Figure 2). This action impedes phosphofructokinase, leading to the accumulation of glucose 6-phosphate, which serves as a substrate for the hexose monophosphate pathway used to maintain GSH levels. Another source of NADPH is the NADP+-dependent isocitrate dehydrogenase. Cardiomyocytes harbor different NADP+-isocitrate dehydrogenase isoenzymes in the cytosol and mitochondrial matrix (50).
Pyruvate converts citrate into isocitrate by inducing aconitase, one of the Krebs Cycle enzymes responsible for this conversion. Consequently, pyruvate carboxylation may contribute to the generation of NADPH, an essential electron carrier for maintaining GSH in its reduced antioxidant state. It's worth noting that the electron processing by NADPH enzyme for malate reduces pyruvate carboxylation through the enzyme and converts this malate to isocitrate, which is then oxidized by NADP+ and isocitrate dehydrogenase. Therefore, pure NADPH doesn't directly contribute to this product.
Instead of converting pyruvate into malate and then citrate, citrate and isocitrate flow out of the mitochondria and enter the cytosol, effectively supplying NADPH via the mitochondrial membrane (Figure 2) (51). Low-flow ischemia and reperfusion, which is characterized by a reduced concentration ratio of GSH to its oxidized form (glutathione disulfide, GSSG) in isolated working guinea pig hearts, diminished the myocardial GSH redox state. Following ischemic pyruvate therapy, GSH/GSSG levels were restored, and the NADPH/NADP+ redox status and citrate content were enhanced (52).
Additionally, pyruvate prevents H2O2 from inactivating oxidative-sensitive enzymes in the open heart, thereby mitigating cardioplegia-induced arrest in the cytoplasmic myocardium and cardiac arrest, as well as cardiopulmonary resuscitation in canine myocardium (53). Bassenge et al. found that pyruvate-induced changes in cytosolic (NAD+)/(NADH) lactate affect the synthesis of RONS by NADH oxidase. Pyruvate concentration-dependent inhibited NADH oxidase activity by 55% in cardiac homogenates. Oxypurinol, a xanthine oxidase inhibitor, did not alter ROS production induced by lactate. Neither pyruvate nor lactate affected RONS formation by a xanthine/xanthine oxidase system in vitro, with pyruvate exclusively suppressing the source of RONS, except for xanthine oxidase.
Upon reperfusion after 5 minutes of acute ischemia in isolated guinea pig hearts, a significant 60-second burst of RONS synthesis occurred. This RONS burst was notably reduced by pyruvate, with 5 mM pyruvate suppressing it by 75%. In contrast, lactate increased post-ischemic RONS generation (54). It's important to note that mitochondrial prevention by the monocarboxylate transport inhibitor cyano-3-hydroxycinnamate (at a concentration of 0.5 mM) did not inhibit the suppression of RONS by pyruvate, whereas sarcolemma pyruvate uptake had no impact. Consequently, in this model, pyruvate's antioxidant actions are primarily cytosolic, unlike its energy-producing and inotropic activities that require mitochondrial metabolism (55).
Pyruvate's antioxidant properties are believed to contribute to its ability to enhance β-adrenergic pathways in shocked myocardium. Ischemia-reperfusion reduced β-adrenergic stimulation of contractile function in isolated guinea pig hearts, but pyruvate therapy largely restored it. The relationship between cardiac output and isoproterenol concentration was altered by ischemia-reperfusion, as evidenced by the drop in EC50 from 0.3 ± 0.06 nM pre-ischemia to 5.2 ± 1.9 nM in anesthetized hearts. However, when 5 mM pyruvate was administered after 15 minutes of reperfusion, the EC50 for isoproterenol decreased to 1.1 ± 0.3 nM. Consequently, 2 nM isoproterenol, which had a pronounced inotropic effect in pre-ischemic hearts, was ineffective in shocked myocardium. Still, 5 mM pyruvate substantially restored the inotropic response to isoproterenol (56).
However, while pyruvate alone increased GATP levels, GATP remained comparable in the absence vs. presence of 2 nM isoproterenol stimulation. The increase in GATP couldn't account for the fivefold greater power of the hearts receiving 2 nM isoproterenol and pyruvate compared to the hearts receiving 2 nM isoproterenol alone. In contrast, the antioxidant ratios GSH/GSSG and NADPH/NADP+ were significantly increased by pyruvate, similar to the effects of the sulfhydryl antioxidant N-acetylcysteine. This increase in antioxidant ratios accompanied the augmentation of isoproterenol-induced power and GSH/GSSG, similar to pyruvate, without altering ΔGATP levels (52). Therefore, pyruvate's antioxidant mechanisms strengthen β-adrenergic inotropic responses in shocked myocardium (57).
Other research demonstrated that inhibiting the activity of Pyruvate dehydrogenase kinase 4 (PDK4) altered the stimulation of glucose oxidation, potentially alleviating glucose oxidation, increasing glucose uptake, and reducing myocardial ischemia-reperfusion injury (58).
3.8. Tannins and Their Biological Effects
Tannins are a diverse group of water-soluble polyphenolic compounds with a high molecular weight (500 - 3000 Daltons) and up to 20 hydroxyl groups. They are commonly found in plants, foods, and beverages (59). Tannins are chemically reactive phenolic compounds that form both inter- and intramolecular hydrogen bonds, enabling them to interact with and precipitate macromolecules such as proteins and carbohydrates. They are also responsible for the astringent taste found in many fruits and vegetables (60). Astringency tends to increase with the presence of one to five hydroxyl groups but decreases when there are seven or more, as steric hindrance begins to counteract the strength of hydrogen bonding (61).
Despite being abundant in human food (with an estimated daily intake of 0.1 - 0.5 g), tannins have received relatively little attention due to their polymeric structure and structural complexity (62). In recent years, there has been growing interest in the potential beneficial effects of proanthocyanidins and their monomers on human health. These effects include immunomodulation, anti-inflammatory properties, anticancer activity, antioxidant effects, cardio-protection, and anti-thrombotic properties (63).
For instance, sumac extracts and isolates containing tannins are known to enhance the quality and oxidative stability of animal products such as meat and milk (64).
However, more research is needed to fully understand the pharmacological and toxicological activities of tannins and their specific roles in human health. Despite the fact that several plant-derived proanthocyanidins are now widely consumed as dietary supplements, there is currently no conclusive evidence regarding their safety or potential for long-term harm (65). In this context, we have examined the most recent research and findings regarding the characteristics of tannins, with a particular focus on the antioxidant effects of proanthocyanidins on the heart (66).
3.9. Cardioprotective Properties
The relationship between proanthocyanidin consumption and its cardioprotective effects has been extensively investigated (67). Numerous in vitro and animal studies have been conducted to elucidate the mechanisms through which proanthocyanidins contribute to the prevention and improvement of various cardiac conditions, including atherosclerosis, blood pressure regulation, and lipid homeostasis (68).
Recent research has identified additional modes of action. For example, oligomeric proanthocyanidins (OPCs) derived from Crataegus oxyacantha L. have been shown to inhibit monocyte-to-macrophage differentiation in atherosclerosis, as demonstrated in both in vitro and in vivo studies. Oligomeric proanthocyanidins were found to reduce levels of vascular cell adhesion protein 1, chemokine CCL2, as well as inflammatory markers like MMP 2 and 9 and PPAR. These findings suggest that these phytochemicals may play a role in modulating macrophage activity during the early stages of atherosclerosis and related conditions (69).
Another animal study investigated the cardioprotective effects of proanthocyanidins in rats with doxorubicin-induced heart damage. Proanthocyanidins significantly reduced doxorubicin-induced electrocardiographic abnormalities and corrected ventricular tachycardia induced by aconitine. Additionally, proanthocyanidins markedly reduced the biochemical changes induced by doxorubicin, including creatine kinase-myocardial band, LDH, MDA, SOD, and CAT levels (70). Moreover, GSPs have been observed to possess antioxidant, anti-inflammatory, and antiapoptotic effects, making them capable of protecting the liver against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress (71).
Furthermore, researchers reported that intraperitoneal administration of fisetin (20 mg/kg) 15 minutes before surgery attenuated I/R-induced mitochondrial dysfunction, restored mitochondrial copy number, modulated Pgc1 gene expression, reduced mitochondrial fission, enhanced mitochondrial fusion, and promoted mitochondrial autophagy. These actions collectively counteracted the I/R-related reduction in mitochondrial mass in renal tissue (72).
There is also evidence suggesting that a diet rich in proanthocyanins can protect against acute ischemic brain injury in rats, leading to improved motor function, reduced cerebral infarction volume, and decreased levels of peroxidative markers such as 4-hydroxyl reduction and advanced glycation products. Additionally, inflammatory markers, including CCL2, ionized calcium-binding adapter molecule-1, and TNF-α, were reduced (73).
Furthermore, a study highlighted the bioactive components of Tamarindus indica, specifically Thymine and 4H-Pyran-4-one, 2,3-dihydro-3,5-dihydroxy-6-methyl-, which exhibited significant binding affinity with the human AT1 receptor antagonist via PPARγ agonist. These antioxidant properties could potentially serve as a dietary supplement for mitigating cardiac toxicity (74).
Lastly, recent research explored the inhibitory effects of procyanidin B2 on the NOD-like receptor family. A significant downregulation of NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs) was observed, leading to the inhibition of caspase-1 activation and IL-1β secretion in response to lipopolysaccharides (LPS). Furthermore, procyanidin B2 reduced the production of reactive oxygen species (ROS) induced by LPS and suppressed the transcriptional activity of activator protein-1 (75, 76).
4. Conclusions
This review has provided a comparative analysis of the antioxidant effects of various plants and tannins in the context of myocardial ischemia. It is evident that different antioxidants possess anti-ischemic and anti-reperfusion properties.
