







1. Background
Primary hyperoxaluria (PH) is an autosomal genetic disorder in which glycosylate metabolism is affected leading to oxalate accumulation in the body (1). There are three common PH types. Type 1 PH is due to a deficiency of alanine glycosylate aminotransferase (AGT). This enzyme acts in hepatic peroxisomes, catalyzing the transamination of glycosylate to glycine concomitant to produce alanine from pyruvate (2, 3). The type 2 PH is resulted from a deficiency in glycosylate reductase/hydroxy pyruvate reductase (GRHPR) enzyme. This leads to aberration in glycosylate reduction to glycolate ending up in oxalate accumulation (2, 3). The type 3 PH is due to deficiency in 4-hydroxy 2-oxoglutarate aldolase mitochondrial enzyme (DHDPSL). This enzyme is responsible for transformation of 4-hydroxy 2- oxoglutarate to pyruvate and glycosylate. In all three PH forms, glycosylate detoxification is defected. Subsequently, glycosylate is transformed into oxalate by cytosolic lactate dehydrogenase (LDH) (2, 3).
Type 1 PH is the most prevalent subtype comprising around 80% of the patients with this condition (4). Nevertheless, determining the exact penetrance of PH is almost problematic as most cases are late-diagnosed or even remain undiagnosed. Overall, the frequency of PH is estimated as 1 to 3 cases per 1 million people with an incidence rate of 1 per 100.000 births. However, higher rates have been detected in some populations being more prevalent in regions with commonplace familial marriages. PH has been noted in 1% of children with end-stage renal disease (ESRD) (5, 6).
Type 1 PH also renders the most dreadful subtype of the disease. Type 1 PH usually presents within the first months of life, inflicting a high mortality rate. However, in some cases, the disease may present within the second decade of life with recurrent kidney stones due to excessive renal oxalate accumulation (7). Furthermore, excessive oxalate may also precipitate in other body organs, such as retina, myocardium, blood vessels, skin, bone, and nervous system (8).
Kidney transplantation as the sole intervention in PH may deliver desired outcomes because of a high rate of nephrocalcinosis recurrence. This unfavorable outcome has limited the kidney transplantation for mild disease manageable by vitamin B6, crystallization inhibitors, and thiazide diuretics (6). In established cases of PH, liver transplantation (LT) provides a more viable and valid therapeutic option. Due to the progressive and irreversible renal failure in patients with PH, concomitant LT, and kidney transplant are inevitable (9). Performing LT before the presentation of kidney failure seems to result in more favorable responses in patients at the early stages of the disease (10).
2. Objectives
The aim of the present study was to assess post-LT survival and complications in patients with PH who underwent LT alone or in combination with kidney transplant.
3. Methods
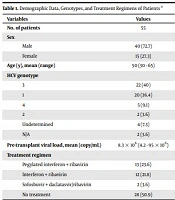
This cross-sectional study was conducted on 18 patients with an established diagnosis of PH who were either liver transplanted or liver and kidney transplanted. The patients were transplanted in the Shiraz Organ Transplantation Center affiliated to the Shiraz University of Medical Sciences, Shiraz, Iran, which is the largest transplantation center of Iran. Also, informed consent was obtained from the patients’ parents.
Demographic information, as well as laboratory and clinical data (pre- and post-transplantation), were obtained by either interviews or reviewing clinical archives. These data included age, sex, weight, type and source of grafts, blood group, sonography findings, graft rejection, post-transplant complications, mortality, and survival rate. The laboratory records included white blood cell counts, hemoglobin, platelet count, blood urea nitrogen, creatinine, glomerular infiltration rate (GFR), hepatic enzymes, albumin, total protein, and coagulation tests. Statistical analyses were performed in SPSS 18 software. Kaplan-Meier curve was used to estimate the survival rate.
4. Results
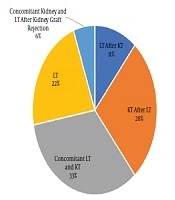
Of 18 studied patients with PH, 12 patients (66.7%) and six patients (33.3%) were male and female, respectively. The age range varied between 3 and 32 years with the mean age of 18.89 ± 7.42 years. The patients’ weight range was 13 - 73 kg, with the mean weight of 47.39 ± 17.18 kg. Blood groups were as A, B, AB, and O with respective frequencies of 3, 5, 2, and 8. Rh D antigen was positive in 17 cases. Concomitant kidney and LT were performed in 6 cases (33%) (Figure 1).

Figure 1.
The frequency of kidney and liver transplantation in 18 patients with primary hyperoxaluria
Polyuria was the most common clinical presentation (11/18). ESRD and hemodialysis were noted in 13 and 12 patients, respectively (Table 1). Basic pre-transplant laboratory parameters are shown in Table 2.
Table 1.Clinical Presentations of the Patients with Primary Hyperoxaluria Pre-Transplant
| Complications | Frequency |
|---|---|
| Chronic renal insufficiency | 18 |
| Hemodialysis | 12 |
| Polyuria | 11 |
| Nephrocalcinosis | 10 |
| Kidney stone | 8 |
| Renal colic | 6 |
| Urinary tract infection | 6 |
| Stone excretion | 4 |
| Hematuria | 4 |
| Renal tubular acidosis | 2 |
| Peritoneal dialysis | 1 |
Table 2.Demographic Information And Pre-Transplant Laboratory Data in 18 Patients with Primary Hyperoxaluriaa
| Parameters | Minimum | Maximum | Values |
|---|---|---|---|
| Age | 3 | 32 | 18.88 ± 7.41 |
| Weight | 13 | 73 | 47.38 ± 17.17 |
| Age at introducing to the waiting list | 2.5 | 28 | 17.6 ± 6.7 |
| White blood cell count, 103/µL | 3.8 | 10.1 | 6.33 ± 2.14 |
| Hemoglobin, g/dL | 8 | 12.3 | 9.77 ± 1.26 |
| Platelet count, 103/µL | 43 | 346 | 180.2 ± 83.31 |
| Blood urea nitrogen, mg/dL | 14 | 102 | 50.38 ± 24 |
| Creatinine, mg/dL | 0.7 | 10.4 | 5.85 ± 2.72 |
| Glomerular filtration rate | 5 | 35 | 15.86 ± 8.09 |
aValues are expressed as mean ± SD.
In post-transplant sonographic findings of the liver, the size and echogenicity were increased in 1 and 5 patients, respectively. Furthermore, six patients showed hepatic hematoma in sonographic findings. Echogenicity of the kidney parenchyma was increased in 4 out of 18 patients. Hepatic arterial thrombosis, biliary complications, infections, and graft rejection were the most frequent transplantation complications (Table 3).
Table 3.Post-Transplant Complications (Liver or Simultaneous Liver-Kidney Transplantation) in 18 Patients with Primary Hyperoxaluria
| Post-Transplant Complications | Frequency |
|---|---|
| Biliary complications | 5 |
| Infections | 5 |
| Graft rejection | 4 |
| Hepatic arterial thrombosis | 4 |
| Ascites | 2 |
| Portal vein thrombosis | 1 |
| Bone marrow suppression | 1 |
| Cardiovascular problems | 1 |
| Renal vein thrombosis | 1 |
| Hepatic vein thrombosis | 0 |
Of 18 patients, seven patients (38.9%) died due to various complications within a year of transplant. Late-presenting complications were not observed in 10 out of 18 patients (Table 4). Post-transplant laboratory and clinical parameters are presented in Table 5.
Table 4.Late-Presenting Complications After Transplantation (Liver or simultaneous Liver-Kidney Transplantation) in 18 Patients with Primary Hyperoxaluria
| Late-Presenting Post-Transplant Complications | Frequency |
|---|---|
| Death due to graft failure | 3 |
| Biliary obstruction | 1 |
| Death due to sepsis | 1 |
| Death due to multiorgan failure | 1 |
| Death due to excessive bleeding | 1 |
| Death due to heart failure | 1 |
| No complication | 10 |
Table 5.Post-Transplant Laboratory Data in 18 Patients with Primary Hyperoxaluriaa
| Parameters | Minimum | Maximum | Values |
|---|---|---|---|
| White blood cell count, 103/µL | 2.3 | 22.1 | 10.04 ± 6.01 |
| Hemoglobin, g/dL | 5.6 | 14 | 9.11 ± 2.36 |
| Platelet count, 103/µL | 42 | 429 | 218.33 ± 95.94 |
| Blood urea nitrogen, mg/dL | 10 | 93 | 41.5 ± 21.6 |
| Creatinine, mg/dL | 0.9 | 6.5 | 2.83 ± 1.97 |
| Glomerular filtration rate | 8.3 | 87 | 44.63 ± 27.76 |
| Aspartate amino transferase, IU/L | 11 | 4690 | 338.47 ± 1125.25 |
| Alanine amino transferase, IU/L | 12 | 910 | 118.29 ± 234.43 |
| Alkaline phosphatase, IU/L | 102 | 2230 | 534.88 ± 506.36 |
| Albumin, g/dL | 2 | 4.8 | 3.15 ± 0.87 |
| Total bilirubin, mg/dL | 0.3 | 13.9 | 2.14 ± 3.7 |
| Prothrombin time (second) | 11 | 47 | 16.1 ± 8.44 |
| International normalized ratio | 0.8 | 14 | 2.57 ± 3.80 |
aValues are expressed as mean ± SD.
Based on the Kaplan-Meier analysis, the survival rate was 61.1% at the end of the study. The mean survival time was 46.25 ± 18.6 months (Figure 2). The patients succumbed to the disease died within 3 to 320 days (the mean of 61.57 days) post-surgery.

Figure 2.
The survival rate of patients with primary hyperoxaluria after transplantation
5. Discussion
As PH is caused by a deficiency of the hepatic enzymes, it seems that liver transplantation can be an appropriate approach to prevent renal insufficiency and ESRD in these patients. However, ethical issues, the risk, and mortality of transplantation complications have limited early LT in these patients. Accordingly, concomitant kidney and LT after ESRD has been considered as the preferred therapeutic approach in patients with PH. It is noticeable that because of the hepatic nature of the disease and deficiency of hepatic enzymes as the triggering element, kidney transplantation is not the definite treatment for PH, and the deterioration of renal function after kidney transplantation is possible. In patients with high-grade CKD and GFR < 15, initial LT, and consequently kidney transplantation is warranted. In these cases, it is possible to conduct hemodialysis and remove excessive oxalate after LT and protect the grafted kidney from precipitated oxalate (9).
The first case of concomitant liver and kidney transplantation in PH was conducted in 1984 for a patient who already had a damaged kidney graft due to re-precipitation of oxalate within the organ (11). The survival rate in adult PH patients underwent isolated kidney transplantation has been 45%, whereas this rate has reported as 64% in those with simultaneous liver and kidney transplant. In children, these rates have been 14% and 76%, respectively, highlighting the importance of concomitant liver and kidney transplantation in children (12, 13).
The systemic presence of calcium oxalate precipitations in other organs along with kidney stones are still probable even years after transplantation. For this reason, patients should be managed by crystallization inhibitors and be advised to consume a high volume of liquid (12). Nevertheless, isolated kidney transplantation may be appropriate in type 2 PH, as a slow-progressing disease. No report of kidney insufficiency has been found due to PH type 3 (9).
In a study conducted in Germany on 13 children with PH, 2 cases with ESRD underwent isolated LT, 4 cases underwent LT before kidney insufficiency, and finally, 7 cases were subjected to concomitant liver and kidney transplantations. One of the cases with ESRD who had been treated with isolated LT and managed by peritoneal dialysis died of peritoneal infection and hepatic abscess 11 months after transplantation. The second patient with ESRD treated with isolated LT also died of cytomegalovirus infection four weeks after the surgery. Among four children with early LT, 3 cases were completely recovered, whereas one patient developed kidney stones and then ESRD after 5.5 years. This patient underwent kidney transplantation 1.8 years later and now has a good clinical condition. Among patients simultaneously transplanted for liver and kidney, they had suitable clinical conditions with only one patient suffering from graft rejection due to steroid resistance. Another patient in this group was infected with polyoma. Overall, it seems that concomitant liver and kidney transplantation renders better therapeutic outcomes in patients with PH. On the other hands, isolated LT in patients with PH complicated by ESRD may not be effective in preventing mortalities. Early LT may also be partially beneficial; nevertheless, these patients may suffer from secondary complications and need kidney transplantation in the future (14).
It has been suggested that early LT precedent to kidney failure can be beneficial in children with slow-progressing PH or those with PH presented during infancy. Nevertheless, simultaneous liver and kidney transplantation is the optimal choice in patients with ESRD (15).
Based on the Cochat et al. (10) study, selecting the type of transplantation in patients with PH can be optimized based on GFR condition. Accordingly, in patients with GFR of 40 - 60, isolated LT is applicable; however, transplantation is not the immediate option in these patients. Among those with GFR of 20 - 40, concomitant liver and kidney transplantation is considered; nonetheless, isolated kidney transplantation is sometimes inevitable in developing countries due to limited facilities and financial resources in these countries. Besides, concomitant liver and kidney transplantation cannot be performed in some patients because of their clinical condition. In patients with GFR < 20, isolated liver or kidney transplantation will be certainly insufficient, and concomitant or consecutive transplantation is the sole therapeutic choice. Also, the simultaneous graft strategy is warranted in PH patients who developed ESRD during the two first years of life (10).
In PH patients who were subjected to kidney transplantation, it is recommended to perform extensive and prolonged dialysis to remove excessive and systemic oxalate from the body before surgery. Furthermore, living donors and close relatives as the source of organs can reduce the risk of graft rejection. Finally, the administration of nephrotoxic drugs, such as cyclosporine should be avoided in these patients (16).
In our study, no patient developed kidney problems post-LT. However, infections occurred in some patients secondary to suppression of the immune system by immunosuppressive drugs. In addition, hepatic arterial and portal vein thrombosis were among common post-surgery incidences. Other post-transplant events were biliary problems observed in 5 patients, of whom one died of biliary obstruction. Such complications highlight the necessity for implementing novel approaches, such as gene therapy, cell therapy, regenerative medicine approaches in this field. Overall, the transplantation was tolerated by ten patients. Nevertheless, the ratio of mortality was relatively high, which should be more scrutinized in future studies.
In our study, we noticed that blood urea nitrogen and creatinine dramatically decreased from the peak averages of 50.39 and 6.59 pre-surgery to 38.67 and 2.42, respectively, which indicated a remarkable improvement in renal function. Also, the mean GFR boosted from 15.86 pre-transplant to 44.63 post-transplant.
All the deaths occurred during the first year post-transplant. This indicates the high risk of mortality early after surgery. However, our study was performed in a relatively short period (a maximum of 6 years), and the mortality rate should be measured over longer periods.
LT can be considered as a definite treatment to avoid renal failure in PH patients. Post-transplant intensive care can escalate the success rate of organ transplantations in patients suffering from PH. Conducting studies over a long period is recommended to evaluate late-presenting complications in these patients.