


1. Background
Fabry disease (FD) is an X-linked disorder of glycosphingolipid catabolism due to absence or deficiency of the lysosomal α-galactosidase A activity (α-GalA, EC 3.2.1.22) (1), which causes the storage of complex glycosphingolipids, mostly globotriaosylceramide (GL-3 or Gb3) and globotriaosylsphingosine (Lyso-GL3), in different tissues (usually in the nervous system, heart, and kidneys). While male patients carrying a genetic mutation develop the disease, female FD patients present wide clinical variabilities, because of the random X-inactivation (Lyon hypothesis) (2).
In FD, a classic form more frequent in male patients without enzymatic activity and a milder non-classic form are described. The classic form usually presents typical signs and symptoms of the disease, such as acroparesthesias, abdominal pain, cornea verticillate, and angiokeratomas. Later in life, these patients develop stroke, hypertrophic cardiomyopathy, and renal failure. In the non-classic or late-onset form, patients are generally less affected, and the manifestations of the disease are usually limited to a single organ, mainly the heart (cardiac variant) or kidneys (kidney variant) (3).
Although FD has been a known pathology for more than 120 years, during the last 20 years, the prognosis has changed because of the availability of enzyme replacement therapy (ERT) (4). FD is pan-ethnic, and its prevalence ranges from 1:40,000 men to 1:117,000 births (5). A recent analysis of some international results of neonatal screening has shown a frequency of 1:22,570 men in classic form and 1:1,390 men in non-classic forms, which would position this pathology as the most frequent lysosomal storage disorder (6).
Kidney failure presents with protein loss in urine and progressive renal impairment (1). The development of FD nephropathy resembles diabetic nephropathy, with glomerular hyperfiltration (GHF) as the first stage, contributing to proteinuria and progressive kidney failure (7). GHF has been indicated in different publications, but its prevalence is poorly understood in Fabry patients.
2. Objectives
To report the prevalence of glomerular hyperfiltration in Fabry disease patients and its association with clinical variables.
3. Methods
Adult patients (≥ 18 years) were evaluated at the moment of FD diagnosis. Plasma α-galA activity was measured using the fluorometric method on filter paper at Dr. Chamoles Laboratory (CABA, Argentina), and the genetic test was performed by multiplex ligation-dependent probe amplification (MLPA) and sequencing at Baylor Genetics (Houston, Texas, USA). The glomerular filtration rate (GFR) was estimated by the CKD-EPI formula (8). Estimated GFR over 125 mL/min/1.73 m2 was considered as GHF (9).
The variables studied were: central nervous system (CNS) compromise, peripheral nervous system (PNS) compromise, presence of arterial hypertension (AHT), cardiac arrhythmia, left ventricular hypertrophy (LVH), albuminuria/proteinuria, cornea verticillata, gastrointestinal involvement, treatment with inhibitors of the renin-angiotensin-aldosterone system (RAAS), deafness, and presence of angiokeratomas.
CNS compromise was determined by clinical stoke history or asymptomatic lesions on brain magnetic resonance (1). PNS compromise was considered in patients suffering from acroparesthesias or in the presence of alterations in quantitative sensory testing (QST) (10).
AHT was defined according to the guidelines of the American College of Cardiology (ACC) and the American Heart Association (AHA) (11). Cardiac arrhythmia was evaluated by the presence of electrophysiological alterations in 12-lead electrocardiogram (ECG) and left ventricular hypertrophy (LVH) by transthoracic echocardiography (1). Septal and left ventricular posterior wall between 6 and 11 mm were considered to be normal (12). Albuminuria was defined as persistent values > 30 mg/24 hours and proteinuria as persistent values > 300 mg/24 hours (13).
Cornea verticillata was assessed by ophthalmological slit-lamp examination. Gastrointestinal involvement centered on abdominal pain and recurrent diarrhea and nausea without any other cause than FD (1). Deafness was determined with tonal audiometry and the presence of angiokeratomas by dermatological exam.
3.1. Data Analysis
SPSS version 20 (IBM Inc., Chicago, Illinois, USA) was used to perform the analysis. The normal distribution of continuous variables was tested with the use of the Shapiro-Wilk test. Descriptive statistics were presented as mean ± standard deviation (SD) for continuous variables and percentages for categorical variables. To compare demographic, clinical, and laboratory data, the chi-square or Fisher’s exact test, ANOVA, Kruskal-Wallis or independent t-test were used. P value of less than 0.05 was considered statistically significant.
4. Results
Forty-eight patients (28 women [58.3%] and 20 men [41.7%]) with a confirmed diagnosis of FD (35.9 ± 11.7 years) were studied. A total of 15 index cases were identified. The α-galA activity was low in all male patients and only in 14.3% of female patients. An overall of 14 GLA gene mutations were found: E398X, L415P, M296V, L106R, R227Q, A292T, c.448.delG, R363H, C382Y, R301Q, D109G, del 3&4 exons, W81X, and R342X; all of which were pathogenic FD mutations (http://fabry-database.org/mutants). 9/48 patients (18.8%) with GHF were detected, which included six females and three males (mean age: 28.8 years). The characteristics of the study population are shown in Table 1.
| Variable | Patients (N = 48), No. (%) |
|---|---|
| Sex | |
| Male | 20 (41.7) |
| Female | 28 (58.3) |
| Age (mean ± SD) | 35.9 ± 11.7 |
| Index cases | 15/48 (31.3) |
| Decreased α-galA activity | |
| Male | 20/20 (100) |
| Female | 4/28 (14.3) |
| Mutations of the GLA gene | 13 |
| Glomerular hyperfiltration | 9/48 (18.8) |
Study Population Characteristics
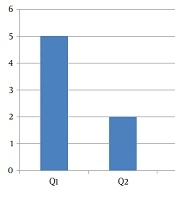
In the first quartile of age, a major prevalence of GHF was evidenced (Figure 1). Figure 2 shows the distribution of GHF frequencies by age. A significant association was found between GHF and CNS compromise (P = 0.021), PNS compromise (P = 0.001), cardiac arrhythmia (P = 0.001), cornea verticillata (P = 0.009), and gastrointestinal involvement (P = 0.009). No significant relationship was noted between GHF and proteinuria (P = 0.859), LVH (P = 0.386), AHT (P = 0.248), RAAS inhibitors treatment (P = 0.248), deafness (P = 0.564), and the presence of angiokeratomas (P = 0.386). Table 2 shows the frequencies and associations between GHF and the different variables studied.
| FD Patients With GHF | FD Patients Without GHF | P Value | |
|---|---|---|---|
| CNS involvement | 2/9 (22.2%) | 14/39 (35.9%) | 0.021a |
| PNS involvement | 7/9 (77.8%) | 29/39 (74.4%) | < 0.001a |
| Cardiac arrhythmia | 0/9 (0%) | 6/39 (15.4%) | < 0.001a |
| Cornea verticillata | 3/9 (33.3%) | 12/39 (30.8%) | 0.009a |
| GI compromise | 1/9 (11.1%) | 14/39 (35.9%) | 0.009a |
| Proteinuria | 6/9 (66.7%) | 33/39 (84.6%) | 0.859 |
| LVH | 1/9 (11.1%) | 20/39 (51.3%) | 0.386 |
| AHT | 1/9 (11.1%) | 19/39 (48.7%) | 0.248 |
| RAAS inhibitors treatment | 2/9 (22.2%) | 18/39 (46.2%) | 0.248 |
| Deafness | 2/9 (22.2%) | 20/39 (51.3%) | 0.564 |
| Angiokeratomas | 2/9 (22.2%) | 19/39 (48.7%) | 0.386 |
Frequencies and Association Between Glomerular Hyperfiltration and the Different Variables Studied

Distribution of glomerular hyperfiltration frequencies by quartiles of age

Distribution of glomerular hyperfiltration frequencies by age
5. Discussion
Nephropathy is a serious complication of FD patients (14). Fabry nephropathy presents with different severity stages with a similar progression rate to diabetic nephropathy, whereby male patients with the classic and untreated form usually develop end-stage renal disease in their fourth decade of life (9). In general, male patients without specific treatment exhibit three phases of FD nephropathy; the first is characterized by GHF, the second by renal involvement with albuminuria and proteinuria, and in the third phase, patients develop severe renal disease with cardiovascular and neurological compromise (5).
GHF is considered an early phenomenon in the sequence of events ranging from intraglomerular hypertension to albuminuria, and subsequently to the reduction of the glomerular filtration. The factors involved in its physiopathology are multiple and not so well known, and include numerous humoral factors such as nitric oxide, prostaglandins, RAAS, atrial natriuretic peptide, reactive oxygen species, etc. (15). The presence of such pathogenic factors has previously been reported in patients with FD (16).
Unfortunately, there is no simple and precise method to measure renal functionality that is feasible in daily clinical practice. Glomerular filtration can be evaluated using exogenous or endogenous markers. Measurements with exogenous substances as nuclear markers or inulin are the gold standard to calculate renal function in FD patients (17). However, these methods are expensive and time-consuming. Creatinine is the most used endogenous marker. The CKD-EPI formula has demonstrated more accurate GFR measurements than other formulas, especially in the higher ranges (8). This formula has recently been validated and may be useful in FD patients (9).
In the present analysis, we have found low plasma α-galA activity in all males and only in 14.3% of females. The lyonization would be responsible for the high percentage of normal enzyme activity in women of our population. This X-chromosome inactivation impacts the natural history of female patients with FD (18, 19). We have identified GHF in approximately one-fifth of our samples. The detection of a major prevalence of GHF in the first quartile of age is coincident with the natural evolution of Fabry nephropathy in patients without specific treatment. A recent retrospective observational study in Italy showed a similar result with a GHF prevalence of 24% in a population of 87 FD patients (20).
Clinical manifestations of the classic phenotype have been described before 7 years of age with neuropathic pain crisis in extremities (typically triggered by physical activity, fever, and temperature changes), post-prandial abdominal pain, and frequent diarrhea. Also, dermatological lesions such as angiokeratomas and hypohidrosis may be present before the second decade of life. Later, hearing loss, tinnitus, vertigo, and ocular involvement usually accompany the finding of podocyturia and proteinuria. After the third decade, cardiac manifestations such as arrhythmias, chest pain, LVH, and stroke can occur (21). In this analysis, we observed a significant association between GHF and CNS and PNS compromise, cardiac arrhythmia, cornea verticillate, and gastrointestinal involvement, most of which are manifestations of the classic FD form. No association was found between GHF and proteinuria, LVH, AHT, treatment with RAAS inhibitors, deafness, and the presence of angiokeratomas.
In Argentina, a recent analysis of young patients with classic FD forms and mild or absent nephropathy showed urinary microRNAs suggestive of kidney fibrosis, even in non-albuminuric cases (22). Riccio et al. reported that GHF may be an early marker of renal involvement in FD patients, even before the onset of signs, symptoms, or laboratory changes (20). Although proteinuria can be considered an indirect marker of GHF, there are situations where the urinary protein loss is due to alterations in the glomerular filtration barrier secondary to a glomerular disease rather than a consequence of the GHF per se. Therefore, we can infer that the proteinuria detected in our population would be related to other mechanisms, such as damage in the glomerular filtration barrier produced by the deposition of glycosphingolipids and not by the presence of GHF.
5.1. Conclusions
In summary, this research has evidenced a higher prevalence of GHF in the younger group and a significant association between GHF and some clinical symptoms of the classic form. However, there was no association between GHF and proteinuria or RASS inhibitors treatment. Nowadays, the physiopathology of GHF in Fabry patients is not well known. Even though further research is needed, it is concluded that other mechanisms than glomerular hyperfiltration, like injury by glycosphingolipids deposit into the filtration barrier, might influence the protein loss of Fabry nephropathy.
