




1. Introduction
CNS is defined as a nephrotic range proteinuria (>40mg/m2/h) starting within the first trimester after birth (1-3). It is a rare kidney disorder with heavy leakage of plasma protein into urine. In most of the patients, this is due to mutations in regulatory or structural genes of the renal filtration barrier located in the glomerular capillary wall (2). CNS may also be part of a more systematic disorder or can be caused by some perinatal infections such as TORCH (Toxoplasmosis, other, Rubella, Cytomegalovirus, Herpes). Genetic defects account for most of the CNS patients, but infections are a possible cause, especially in developing countries (3). Different associations between CNS and other organ anomalies have already been reported. The evaluation of possible coexisting malformations is important. These include neurological abnormalities (Golloway-Mowat), ocular defects (mutation in LAMB2 gene), and genital disorders (mutation in WT1 gene) (2, 4). Here, we report a case of congenital nephrotic syndrome and vesicoureteral reflux presenting with urosepsis.
2. Case Presentation
A 25-day-old male newborn, who was the result of a normal vaginal delivery, was referred to our hospital with generalized edema. The pregnancy course was uneventful. However, a large placenta was reported. The primary laboratory tests showed massive proteinuria, hypoalbuminemia, and hyperlipidemia. The results of the investigations of congenital infections were negative. His parents were cousins with no significant health problems. A detailed family history revealed that the first child of this family was also diagnosed with CNS and had died because of dialysis complications when 20 months old. Conservative management with albumin infusion, ACE-inhibitor, and aspirin with anti-platelet dosage was started, but the parents refused nephrectomy.
The baby, then four months old, was admitted to the hospital with high fever and poor feeding.
Sepsis workup was performed. Urinalysis revealed significant pyuria. Urine culture was positive for E. Coli with more than 100/000 CFU. A diagnosis of the urinary tract infection was established.
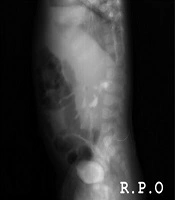
Intravenous antibiotic therapy continued for a period of 14 days. A voiding cystourethrography was performed after cleaning the urine of infection, which showed moderate bilateral VUR (Figures 1 and 2). The fluoroscopical study of the urethra appeared to be normal.

Figure 1.
AP view of VCUG shows moderate bilateral VUR

Figure 2.
R.P.O view shows bilateral moderate VUR
3. Discussion
Resistant to treatment, nephrotic syndrome, and congenital anomalies of the kidney and urinary tract (CAKUT), including ureter pelvic junction obstruction and vesicoureteral reflux, are two important causes of renal impairment in children. Although the etiology of CAKUT is probably multifactorial, it is likely that many causes of CAKUT have a genetic base. There is new growing evidence that different renal and urological malformations may even be caused by gene mutation (5). On the other hand, the genetic base of CNS and some progressive forms of nephrotic syndrome like focal segmental glomerulosclerosis (FSGS) has lately been approved (6). Mammalian kidneys originate from the two compartments of embryonic mesoderm (ureteric bud and metanephric mesenchyme), whose interaction between these parts differentiated metanephric mesenchyme to nephron epithelia (5). Failure of this mechanism is considered as one of the main causes of a wide range of renal malformations. Several genes (PAX, SOX, WNT, and others) play an important role during the differentiation process, leading to the formation of nephron epithelia (5, 6). Therefore, the etiology of CAKUT is probably multifactorial (5, 7). Complicated nephrotic syndrome and CAKUT, including ureteropelvic junction obstruction, vesicoureteral reflux, hypoplastic and dysplastic kidney, are important causes of renal impairment in children, and it is likely that many of CAKUT causes are genetic. There is new growing evidence that different renal and urologic malformations may be caused by the same types of gene mutations (6). Only a few studies that support the association between nephrotic syndrome and urologic anomalies have been published. For the first time in 1972, William and his coworkers report seven children out of 21 patients who were admitted due to nephrotic syndrome and were found to have congenital anomalies of the genitourinary tract; they brought up the need for urological evaluation of all patients with nephrotic syndrome. However, they did not perform any genetic study at that time (8). After that, in 1973, Berger et al. (9) reported transient obstructive uropathy associated with nephrotic syndrome. In 2008, Seyedzadeh et al. (10) reported a case of Pfeiffer syndrome (acrocephalosyndactyly) with high-grade bilateral vesico-ureteral reflux and discussed the importance of studying urologic problems in patients with genetic diseases, especially if there are urinary symptoms. Here we report vesico-ureteral reflux in a four-month-old patient with congenital nephrotic syndrome. Owing to the definitive role of genetic mutation in the pathophysiology of CNS, the detection and presence of vesico-ureteral reflux suggest the existence of a common source of genetic disorder in this association.
Therefore, according to our findings, we recommend that patients with CNS should be considered and evaluated for probable association with urinary anomalies, although more case studies along with genetic evaluation are recommended.