

1. Introduction
Rare soft-tissue sarcomas in the extremities, known as malignant peripheral nerve sheath tumors (MPNSTs), are frequently linked to neurofibromatosis (1). Up to 50% of MPNSTs develop in individuals with neurofibromatosis type 1 (NF-1), 10% of MPNSTs are radiation-induced, and 40% are sporadic. The MPNSTs make up 5% - 10% of all soft-tissue sarcomas (2). Adults aged 20 - 50 frequently have MPNSTs, typically associated with the main nerve trunks (3). Most are aggressive, frequently recur, and frequently develop distant metastases (4). The most typical location for metastasis is the lungs (5). The MPNST of the kidney is relatively uncommon (6). We report a rare case of an MPNST of the kidney so that it can enrich the existing medical literature.
2. Case Presentation
An intermittent, dull ache in the right flank that had been present for three months in a 23-year-old man without any known medical conditions was described as having an insidious onset and being non-radiating or referred. The patient also had haematuria, which was on and off for two months and had increased in the last 15 days, associated with the passage of thread-like clots. The patient had a history of fever, which was low-grade, undocumented, and rose over the last four days.
On physical examination, a large abdominal mass of 5 × 4 cm with firm consistency was found, which occupied the right lumbar quadrant with the fullness of the right renal angle. Serum LDH was raised (358 units/L). Urine cytology was negative for malignant cells, and routine urine microscopy suggested 50 - 60 RBCs/HPF. Ultrasound of the whole abdomen showed a large, moderately well-defined heterogeneous echotexture lesion (94 × 87 mm) in the upper-mid pole of the right kidney with internal vascularity and mass effect over the ipsilateral renal pelvis causing moderate hydronephrosis.
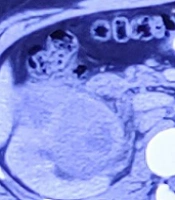
Contrast-enhanced computed tomography (CECT) of the chest and whole abdomen showed the size of the right kidney to be 11.2 × 5.7 cm with a heterogeneously enhancing lobulated mass (9.2 × 6.4 × 7.6 cm) arising exophytically from the anterior cortex of the upper and mid poles of the kidney with internal non-enhancing areas suggestive of necrosis. There was adjacent perinephric fat stranding with no evidence of intralesional fat or increased vascularity. The lesion was hypo-enhancing and was compressing the pelvicalyceal system causing mild hydronephrosis. Superolaterally, it was abutting the inferior surface of the right lobe of the liver with a maintained fat plane. Medially, it was abutting the second part of the duodenum with indistinct fat planes. It was closely abutting the inferior vena cava (IVC) with maintained fat planes (Figure 1). Posteromedially, it was abutting the right psoas muscle with indistinct fat planes. No thoracic metastasis was observed.
 showing right renal vein dilated with heterogeneous attenuation in the lumen (tumor thrombus) with normal inferior vena cava (IVC)")
Abdomen contrast-enhanced computed tomography (CECT) showing right renal vein dilated with heterogeneous attenuation in the lumen (tumor thrombus) with normal inferior vena cava (IVC)
After preoperative anesthetic clearance, the patient underwent an open radical nephrectomy. Intraoperative findings included a right renal mass of 12 × 8 × 5 cm adhered to the ascending colon and duodenum, abutting liver. Deep vein thrombosis (DVT) prevention was advised post-operatively, along with antibiotics and painkillers. Since then, the patient has received routine follow-ups every three months.
The histological assessment of the biopsy specimen was suggestive of an MPNST. The mass had wavy nuclei in an eosinophilic cytoplasm and spindle-shaped cells organized in bundles and fascicles (Figure 2). The specimen margins were microscopically clear of tumor cells. S-100 and Vimentin were stained positive for MPNST (Figure 3).

Clockwise from top left: Haematoxylin-eosin staining showing the geographic pattern of necrosis, tumor cells arranged in sheets, and ovoid and spindle-shaped cells with hyperchromatic nuclei

Clockwise from top left: Slides showing S-100 stained positive, Vimentin stained positive, and CD56 stained positive
3. Discussion
The MPNSTs are sporadic or radiation-induced aggressive soft-tissue sarcomas that are uncommon and linked to neurofibromas in NF-1 (1). Patients with NF-1 typically appear as a single, rapidly growing, painless mass, several rapidly enlarging masses, or a new onset of pain related to pre-existing plexiform neurofibromas (2). The most frequent parts in which MPNSTs occur include the head and neck area, trunk, and extremities (7). There is rarely kidney involvement. Clinically, it can be challenging to tell various kidney sarcomas apart from the MPNST kidney.
For diagnosis and grading, histopathological and immunohistochemical tests are required (8). The MPNSTs are big (> 5 cm) and tan and grey, with a fusiform appearance. They display the characteristics of spindle cells under a microscope, including a fascicular pattern, varying degrees of mitosis, necrosis, and tumor calcification (9). Benign neurofibroma or schwannanian cells that have not undergone a malignant change could be found. S-100 protein, neuron-specific enolase, and other proteins are detected in MPNSTs (10). In this case of kidney lesion, WT1 staining was performed to rule out Wilms’ tumor (11). Synaptophysin and chromogranin staining was completed to rule out neuroendocrine tumors (12), CK7 and panck for ruling out carcinoma (13), desmin, SMA, and CD34 for ruling out endothelial tumors (14), CD10 for ruling out RCC (15), HMB45 staining for ruling out malignant melanoma (16), and TLAB3 and BcL2 for ruling out synovial tumors (17).
Computerized tomography and magnetic resonance imaging (MRI) are useful imaging methods for determining the size of the tumor and may suggest where the tumor originated (18). Increased uptake on a positron emission tomography scan and the evidence of heterogeneity, necrosis, and bleeding in an MRI could both be used to identify malignant transformation. The three most crucial prognostic markers for MPNSTs are tumor size, local recurrence, and the extent of surgical resection. The only option for treating MPNSTs is a complete surgical resection, which offers the only chance for recovery (19, 20). The MPNSTs are regarded as cancers resistant to radiation and chemotherapy. However, postoperative radiation, advised by an oncology consensus group, plays a part in both disease-free and overall survival (20). The prognosis is still dismal despite multimodal therapy, which includes vigorous surgical resection and adjuvant radiation (19).
3.1. Conclusions
Malignant tumors of the kidney, called MPNSTs, are uncommon. Wide local excision, if possible, has been found to be the best course of action because the prognosis heavily depends on how thoroughly the tumor is surgically removed. It is advised to have postoperative radiation.
