





1. Context
The solitary kidney, whether congenital or acquired, is significant from a nephrologist’s perspective due to its reduced nephron count compared to individuals with two kidneys. Patients with congenital solitary kidneys (CSKs) represent a specific group of children often associated with congenital anomalies of the kidneys and urinary tract (CAKUT). A substantial proportion of solitary kidney patients remain at risk for chronic kidney disease (CKD) progression. However, distinguishing patients at high risk for CKD progression from those at lower risk presents significant challenges. Currently, there are no established guidelines or consensus for the follow-up and management of patients with acquired solitary kidneys regarding long-term outcomes.
1.1. Embryology of the Kidney
Normal kidney development begins in the third week of gestation (1). During the fourth week, the urogenital ridge forms, leading to the development of the nephrogenic cord and gonadal ridge (2). The nephrogenic cord gives rise to the pronephros, mesonephros, and metanephros. The mesonephros forms the mesonephric duct (Wolffian duct). In the fifth week, the metanephric blastema, derived from the intermediate mesoderm, induces the mesonephric duct to develop an outgrowth known as the ureteric bud (UB) (2). In females, the mesonephric duct degenerates, whereas in males, it develops into the epididymis, vas deferens, seminal vesicles, and ejaculatory duct (2). The Mullerian ducts (paramesonephric ducts) develop into the uterus, fallopian tubes, proximal third of the vagina, and cervix in females (2).
During the sixth week of gestation, the UB undergoes a branching cascade, forming the collecting tubule and basic renal architecture, including the renal pelvis, cranial and caudal lobes of the kidney, and major calyces (2). By the seventh week, the formation of minor calyces occurs from the UB, and multiple blastemal caps are formed (2). These blastemal caps develop into nephric vesicles, which give rise to nephric tubules. The nephric tubule consists of an S-shaped Bowman capsule, proximal tubule, distal tubule, and loop of Henle (2). Nephrogenesis continues until the 36th week of gestation (1, 3), after which no new nephrons are formed.
Children born before 36 weeks of gestation may have fewer nephrons. Before the sixth week, the kidneys are located in the sacral region and ascend to the lumbar region between the sixth and ninth weeks of gestation (2). Low birth weight and prematurity are indicative of low nephron numbers, which are considered risk factors for hypertension in middle-aged adults (1). Preterm birth (before 37 completed weeks of gestation) and extreme preterm birth (before 28 completed weeks of gestation) are considered risk factors for CKD from childhood to adulthood (1).
2. Evidence Acquisition
We conducted a review of the PubMed, Google Scholar, and Science Citation Index databases. Our review included retrospective and prospective studies that described the underlying causes, outcomes, and follow-up of patients with congenital and acquired solitary kidneys. Cross-sectional studies were excluded. The available data were analyzed to determine the effects of lifestyle modifications, nutritional interventions, and pharmacological treatments on the short- and long-term outcomes of patients who have undergone unilateral nephrectomy.
3. Results
3.1. Congenital Solitary Kidney
The anatomical or functional absence of one kidney at birth is known as CSK (4). This condition arises when there is an embryonic insult leading to the absence of the nephrogenic cord (nephrogenic ridge) or failure to form the UB, resulting in abnormal kidney development (5). The CSK may be associated with other structural anomalies derived from the mesonephric duct or Wolffian duct in males and the Müllerian duct in females. Genital anomalies are 3 to 4 times more frequent in females compared to males (5). Common male genital anomalies include those of the vas deferens, seminal vesicles, and epididymis (5). In females, commonly associated genital anomalies include unicornuate uteri without an intact ipsilateral horn, fallopian tube, or bicornuate uterus (5).
The CSK occurs in approximately 1 in 1000 births, more commonly on the left side, with a male predominance ratio of 1.8:1 (male to female) (6). Approximately one in three cases of CSK is related to CAKUT (4). Vesicoureteral reflux (VUR) is the most common urologic abnormality associated with CSK (4). The adrenal gland is absent in only 10% of CSK cases due to its different embryological origin (5). The congenital functional absence of one kidney may be due to aplasia, agenesis, or multicystic dysplastic kidney (MCDK) (4). The MCDK is characterized by a lobulated renal contour, pelvis, parenchyma, and multiple non-communicating cysts (4).
If no additional abnormalities are associated with CSK, compensatory enlargement or growth of the CSK occurs. This compensatory enlargement begins during gestation (around the 20th week, as detected by ultrasonography) and continues until adolescence (4). The exact mechanism of CSK growth remains unknown; therefore, the term "compensatory enlargement" is preferred over "compensatory hypertrophy".
Thirty years ago, Brenner et al. (as cited by Westland et al.) proposed the hyperfiltration hypothesis based on renal mass reduction experiments in rats, observing that a reduction in the number of functional nephrons leads to compensatory enlargement of the remaining nephrons and glomerular hypertension (7). Animal studies have confirmed that CSKs have a compensatory increase in nephron number, although the total number of nephrons remains lower compared to two kidneys (7). Keller et al. (as cited by Westland et al.) supported the hypothesis of glomerular hyperfiltration, describing that patients with primary hypertension had increased glomerular volume and fewer nephrons compared to those without primary hypertension (7). Van Vuuren et al. (as cited by Westland et al.) studied nephron number at 26 weeks of age (adulthood) in pigs with unilateral renal agenesis (URA), reporting 75% of nephrons compared to two kidneys (7). Douglas-Denton et al. (as cited by Westland et al.) studied nephron number on days 27 - 34 (infancy) in sheep that underwent in utero nephrectomy, reporting 72% of nephrons compared to two kidneys (7). Larsson et al. (as cited by Westland et al.) studied nephron number on days 55 - 56 (adulthood) in rats that underwent nephrectomy during the postnatal period, reporting 52% of nephrons compared to two kidneys (7).
The solitary kidney holds particular significance from a nephrologist’s perspective, as it contains fewer nephrons compared to an individual with two kidneys. If a healthy person has 100% of nephrons in both kidneys, a person with a surgically acquired single kidney would have a reduced number of nephrons (50%) compared to CSK (75% of the nephrons of two kidneys) (3).
3.2. Chronic Kidney Disease Progression Among Children with Solitary Kidney
Given the reduced number of nephrons, there is a risk of kidney damage manifesting as proteinuria due to hyperfiltration, hypertension, and a decline in glomerular filtration rate (GFR) over time. Approximately one-third of adults with CSK exhibit proteinuria, arterial hypertension, and impaired kidney function (3). Hutchinson et al. analyzed renal function in children with a congenital solitary functioning kidney by reviewing 45 previously published studies. They reported that 8.4% of children showed decreased renal function, 10.1% developed proteinuria (compared to a reported prevalence of 5 - 15% in the general pediatric population), and 7.4% developed hypertension (compared to a reported prevalence of 2 - 4% in the general pediatric population) (8-11). Sanna-Cherchi et al. (as cited by Westland et al.) conducted a longitudinal study in solitary kidney patients with CAKUT and found that 20% - 50% required renal replacement therapy by age 30 (7). Compared to a reference group, the presence of VUR further deteriorated renal outcomes (hazard ratio, 7.50; 95% confidence interval, 2.72 to 20.68) (7). Argueso et al. (as cited by Westland et al.) conducted a retrospective study on patients with URA and found that 13% developed impaired GFR, 47% had hypertension, 19% had proteinuria, and 4% died of kidney failure (7).
The Kidney of MONofunctional Origin (KIMONO) study, a prospective longitudinal follow-up conducted in the Netherlands, included 400 children with congenital and acquired solitary kidneys. It observed that a decline in GFR typically occurs after age 9, while microalbuminuria develops around age 16 (7, 12). Early progression to renal injury was observed among patients with ipsilateral CAKUT compared to those without CAKUT (12.8 years versus 15.9 years; P < 0.01). The prevalence of CKD was 4% in CSK cases versus 9% in acquired solitary kidney cases (P = 0.05) (7). Kim et al. studied solitary kidney and CKD risk, analyzing cause-specific solitary kidney. The adjusted hazard ratio (95% confidence intervals) for CKD, comparing unilateral nephrectomy to control, was 6.18 (2.31 - 16.49), and for CSK to control, it was 2.22 (0.83 - 5.92), concluding that a solitary kidney independently increases the risk for CKD development (13).
Some patients with CSK already have associated CAKUT or kidney hypodysplasia. In these patients, compensatory nephron increase and kidney enlargement are less likely compared to those without CAKUT or hypodysplasia (7). In CSK, children without compensatory kidney enlargement and/or with ipsilateral CAKUT are at risk of GFR decline and CKD progression, while those without ipsilateral CAKUT and with compensatory kidney enlargement are at lower risk of GFR decline (14-18).
3.3. Role of Genetic and Environmental Factors
Many genetic mutations and environmental factors contribute to CAKUT and CSKs. Most CSKs result from CAKUT, including URA, MCDK, and renal hypodysplasia. Mutations in over 40 monogenic genes are responsible for CAKUT (7, 19, 20). Ten percent of cases exhibit familial aggregation with incomplete penetrance and predominantly autosomal dominant inheritance. However, X-linked and autosomal recessive patterns have also been reported (7). The PAX2, HNF1B, and DSTYK genes are commonly involved in non-syndromic CAKUT, with 10 - 20% of patients developing kidney malformations due to heterozygous mutations in these genes (7). Twenty percent of newborns with CAKUT have chromosomal abnormalities (7). Sanna-Cherchi et al. (as cited by Westland et al.) studied 522 children and identified 72 different copy number disorders in 17% of patients, manifesting as duplications and sub-microscopic deletions leading to kidney hypodysplasia (7). Sander Groen in Woud et al. (as cited by La Scola et al.) studied gene-environment interactions (GxE) in the etiology and pathophysiology of CSK, highlighting the significant effect of maternal obesity and its interaction with the rs3098698 variant (4). Uncontrolled gestational diabetes mellitus and certain drugs (aminoglycosides, antiepileptic drugs, ACE inhibitors, and dexamethasone) can disrupt renal development during the antenatal period (7). The NSAIDs and aminoglycosides are the most common drugs affecting nephrogenesis (7). Although the reason is unclear, observational studies report a left-side and male predominance for CSK.
3.4. Management of Congenital Solitary Kidney
An ultrasound scan using the Doppler function can differentiate between CSK and MCDK during the 18th to 22nd weeks of gestation, as there is a complete lack of blood flow to MCDK. However, MCDK can involute completely after 29 weeks of pregnancy and become undetectable on ultrasound (4). A Technetium-99m DMSA (dimercapto succinic acid) renal scan can further differentiate the existence of a rudimentary kidney (MCDK versus severe hydronephrosis) after childbirth (4). The diagnosis of VUR in a child can be confirmed using fluoroscopic contrast voiding cystourethrography (VCUG). It is recommended that children with CSK be followed until adulthood (4). Genetic analysis is recommended in cases of CSK with extra-renal malformations to diagnose any possible syndrome (4). Ultrasound scans should be performed yearly up to three years of age and then every five years thereafter. In girls, an abdominopelvic ultrasound scan should be performed between thelarche and menarche (21). Proteinuria assessment should be performed yearly, and office blood pressure (BP) measurement is recommended yearly after age 3 (22-25). Creatinine-based estimated GFR (eGFR) evaluation is recommended annually in medium-risk children (without compensatory enlargement of the kidney) with CSK (4). The Schwartz equation can be used to estimate GFR in children with a solitary kidney (7). After one year of age, renin-angiotensin-aldosterone system (RAAS) inhibitors should be used to control BP and treat proteinuria in children with CSK (26-28). Dehydration and nephrotoxic drugs should be avoided in children with CSK (4). The recommended protein and salt intake in children with CSK should match the dietary recommendations for children of the same age and sex (4). Excessive protein and salt intake should be avoided (4). There is no restriction on sports participation for children with CSK, but children and their parents must be aware of the risk of collision injury that may cause blunt kidney trauma (4).
3.5. Acquired Solitary Kidney
Acquired solitary kidneys are observed in individuals who have undergone unilateral radical nephrectomy due to kidney donation, kidney tumors, renal trauma, severe emphysematous pyelonephritis, or an infected destroyed kidney (6). After unilateral nephrectomy, the absolute risk of developing end-stage renal disease (ESRD) remains low, but the relative risk is 3 - 5 times higher (6, 29).
3.6. Pathophysiological Alterations in Solitary Kidney
In healthy kidney donors younger than 60 years, there is a compensatory increase in GFR up to 65 - 70% of the pre-donation GFR (6). Changes occur in renal hemodynamics and nephron structure, including hypertrophy and hyperplasia (6, 30). Long-term follow-up of living kidney donors has shown that an increased glomerular ultrafiltration coefficient (kf) and increased renal plasma flow are responsible for glomerular hyperfiltration and hypertrophy rather than intraglomerular hypertension (31-33). These changes lead to an increase in single nephron GFR (SNGFR), ultimately resulting in glomerular hyperfiltration. Activation of several pathways, including transforming growth factor-beta, interleukin-10, and mammalian target of rapamycin complex (mTOR), leads to compensatory glomerular hypertrophy and CKD progression (6, 31). Patients with CKD with two kidneys have different pathogeneses than kidney donor patients with a single kidney (32). Glomerulonephritis and genetic predispositions may contribute to faster CKD progression in these patients (32).
3.7. Outcomes of Solitary Kidney
Children born with a solitary kidney may develop focal segmental glomerulosclerosis (FSGS) and a progressive decline in GFR (34). However, some long-term follow-up studies of patients undergoing unilateral nephrectomy did not show a progressive decline in GFR (34). Baudoin et al. (as cited by Matas and Rule) studied patients aged 18 - 56 years who had undergone unilateral nephrectomy in childhood and reported that kidney function was maintained; however, patients followed for more than 25 years showed increased proteinuria, high BP, and a higher incidence of renal failure (34). Living kidney donors undergo extensive screening before donation, which may contribute to better outcomes among patients with acquired solitary kidneys (7).
3.8. Living Kidney Donation Outcomes
The long-term renal outcomes of acquired solitary kidney depend on the age at onset, lifestyle, dietary habits, residual nephron mass immediately post-nephrectomy, duration of solitary kidney, and associated comorbidities. Acquired solitary kidney (unilateral nephrectomy due to donation or post-trauma) is usually seen in young or middle-aged individuals, while malignancies are common causes of unilateral nephrectomy in older individuals with associated comorbidities, increasing the risk of CKD progression. Garg et al. (as cited by Matas and Rule) compared 1,278 living donors with 6,359 controls and found that hypertension was more frequent among living donors (16.3%) compared to controls (11.9%) after six years of follow-up (34). Living kidney donors can develop proteinuria over time. A meta-analysis of 48 studies including 5,048 donors reported an average 24-hour proteinuria of 154 mg/day after an average of seven years post-donation, with an average GFR of 86 mL/min (6). Kidney disease improving global outcomes (KDIGO) guidelines recommend assessing albuminuria in living kidney donors at least once a year (6). Using administrative data, Lam et al. (as cited by Matas and Rule) compared 604 living kidney donors with 2,214 controls and observed an increase in eGFR of 0.35 mL/min/1.73 m2 per year in living donors after seven years of median follow-up, while controls had a significantly decreased eGFR (-0.85 mL/min/1.73 m2 per year) (34). Recently, seven studies conducted in six different countries showed no difference in cardiovascular mortality between living donors and matched controls (34). From October 16, 1987, to November 3, 2020, data from the US Scientific Registry of Transplant showed that 633 living donors (0.4%) developed ESRD (34). According to Wainright et al. (as cited by Matas and Rule), the risk of developing ESRD in living donors was significantly higher among recipients’ first-degree relatives (maximum in identical twins) than among unrelated living donors (34). There is evidence of living donors surviving for more than 50 years. However, longer follow-up studies among living donors, including the University of Minnesota series and Canadian series, reported a mean interval of 27 years from donation to the development of ESRD (30). Keys et al. studied 66 living donors who donated 50 or more years earlier and reported that 22 (33.3%) were still alive (35). Sanchez et al. (as cited by Matas and Rule) studied risk factors for CKD onset in living donors at the time of kidney donation and reported that smoking, higher BMI, older age, hyperlipidemia, and high systolic and diastolic BP were associated with the development of proteinuria and CKD later on in living donors (34). African American individuals and the APOL1 genotype may be associated risk factors for the development of CKD among living donors in the long term, as in the general population (34). Pregnancy causes anatomical and physiological changes in the kidney, such as an increase in size and GFR. Ibrahim et al. (as cited by Matas and Rule) reported that risks for the development of hypertension and preeclampsia were higher in pregnancies after donation compared to pregnancies before donation (34). This increased risk of preeclampsia is equivalent to that observed in females with high BMI, gestational diabetes, and chronic hypertension (34). Therefore, living donors of childbearing age must be aware and informed that pregnancies after donation are considered high risk.
An average 11-year follow-up study of 125,427 living kidney donors showed that glomerulonephritis was the leading cause of ESRD in the first ten years after kidney donation, whereas after ten years, diabetes mellitus and hypertension were common causes of ESRD among living kidney donors (32, 36). The major cause of ESRD in living kidney donors appears to be environment-related renal diseases or immunological factors rather than hyperfiltration-induced FSGS and CKD progression (6, 32).
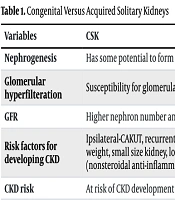
Seun Kim et al. studied deceased donor recipients and living donor recipients, reporting a three-year better outcome in living donor recipients and lower proteinuria levels in deceased donor recipients where donor kidney weight was higher than recipient body weight (37). Young et al. compared living kidney donors with non-donor controls and found that higher levels of serum intact fibroblast growth factor 23 (FGF-23), renal tubular fractional excretion of inorganic phosphorus, lower serum calcitriol, and lower serum phosphorus levels might indicate early onset CKD among living kidney donors (38). Therefore, evidence suggests an overall increased risk of CKD progression among living kidney donors, although the absolute risk remains small. A solitary functioning kidney should not be considered harmless. The distinguishing features of congenital and acquired solitary kidneys are shown in Table 1 (7, 13, 39).
| Variables | CSK | Acquired Solitary Kidney |
|---|---|---|
| Nephrogenesis | Has some potential to form new nephrons. | Nephrogenesis has stopped after nephrectomy. |
| Glomerular hyperfilteration | Susceptibility for glomerular hyperfilteration | Higher susceptibility for pronounced glomerular hyperfilteration |
| GFR | Higher nephron number and higher GFR | Lower nephron number and lower GFR |
| Risk factors for developing CKD | Ipsilateral-CAKUT, recurrent urinary tract infection, prematurity, low birth weight, small size kidney, low socioeconomic status, and nephrotoxic drugs (nonsteroidal anti-inflammatory drugs and aminoglycosides) | Higher age, diabetes, cardiovascular diseases, higher serum uric acid level, metabolic syndrome, unilateral nephrectomy for renal tuberculosis, and nephrotoxic drugs |
| CKD risk | At risk of CKD development | Higher risk of CKD development |
Congenital Versus Acquired Solitary Kidneys
3.9. Management of Acquired Solitary Kidney
Glomerular hyperfiltration may develop one week post-nephrectomy and can continue for more than 10 years in individuals with a solitary kidney (6). The eGFR equations are preferred for monitoring kidney function in living kidney donors (6). However, there are limitations to using eGFR equations, and measured GFR (using iothalamate, iohexol, or diethylenetriamine pentaacetic acid) provides a more accurate assessment for better clinical decision-making (6). The diet should follow the dietary approaches to stop hypertension (DASH) guidelines, which include complex carbohydrates such as fruits, vegetables, legumes, whole grains, low animal-based protein, and low processed food (6). A high-protein diet causes afferent arteriolar dilation, leading to intraglomerular hypertension and proteinuria (31).
Low dietary protein intake (0.6 to 0.8 g/kg/day) is recommended for CKD patients with eGFR < 45 mL/min/1.73 m2 body surface area or proteinuria > 3000 mg/day (40). The nephroprotective effect of low dietary protein has not been observed in any human study involving post-nephrectomy patients (40). A common consensus is to avoid dietary protein intake > 1 g/kg/day for healthy kidney donors, but professional bodybuilders and athletes may be exceptions (6). The recommended dietary sodium intake should be < 4 g/day for individuals at high risk of developing future CKD and for those with a solitary kidney (40). Obese kidney donors have a 1.86 times higher risk of developing ESRD during a 20-year follow-up period post-nephrectomy than non-obese donors (41, 42). In non-bodybuilders and non-athletes with solitary kidneys, the recommended BMI is < 30 kg/m2 (6).
Individuals with solitary kidneys should maintain adequate hydration. Low fluid intake may be associated with increased vasopressin secretion, which can cause increased urinary albumin excretion. For persons with a solitary kidney and eGFR > 60 mL/min per 1.73 m2, the recommended fluid intake should be > 2.5 L/day (6). Smoking is a known risk factor for CKD progression. There was a 5.3 times higher adjusted mortality risk among smoking living kidney donors during a four-year follow-up period compared to non-smoking living kidney donors (43). Smoking should be avoided in individuals with solitary kidneys. There is no specific recommendation for target BP in living kidney donors; however, maintaining a target BP of < 130/80 mm Hg appears appropriate for healthy living kidney donors (6).
Despite limited evidence in humans with solitary kidneys, the renoprotective effect of renin-angiotensin system (RAS) blockers has been demonstrated in animals with solitary kidneys and post-nephrectomy animals (44). The RAS blockers dilate efferent arterioles more than afferent arterioles, resulting in lower intraglomerular pressure and decreased proteinuria. Recently, Ralph et al. investigated the benefits of a potassium-alkali enriched diet and the mechanism of proteinuria in post-nephrectomy patients, reporting improvements in hypertension and acid clearance. They also observed that post-nephrectomy proteinuria might be due to an increase in SNGFR along with the downregulation of megalin (decreased fractional protein endocytosis) (45). Key findings related to risk factors, outcomes, and management of solitary kidneys are shown in Box 1.
| Key Findings |
|---|
| The solitary kidneys (both congenital and acquired) have reduced number of nephrons and are at risk of hyperfiltration induced proteinuria and CKD progression. |
| Compensatory enlargement of CSK begins at 20th week of gestation and continues till adolescence. It occurs due to hypertrophy or increase in number of existing nephrons. |
| Children without compensatory enlargement of kidney and/or with ipsilateral CAKUT are at risk of decline in GFR and progression of CKD. |
| Genetic factors, uncontrolled gestational diabetes mellitus, and drugs exposure (most common NSAIDS and aminoglycosides) can disturb nephrogenesis and renal development in antenatal period leading to occurrence of CSK. |
| Acquired solitary kidneys have lower GFR and higher risk for development of CKD in comparison to CSK. |
| Living kidney donors have increased glomerular kf and high renal plasma flow that leads to glomerular hyperfiltration and hypertrophy rather than intraglomerular hypertension. |
| Glomerulonephritis and genetic predispositions may contribute to faster progression of CKD among living kidney donors. |
| There are increased risks of hypertension and preeclampsia in pregnancies among living kidney donors of child bearing age group. |
| A high-protein diet and high salt intake may further aggravate glomerular hyperfiltration. Excessive protein and salt intake should be avoided in solitary kidney patients. |
| Nephrotoxic drugs, dehydration, and NSAIDS should be avoided in solitary kidney patients. |
| It is recommended that children with CSK should be followed till adulthood. |
| After one year of age, RAAS inhibitors should be used to control BP and treat proteinuria in solitary kidney patients. |
| Avoidance of smoking, DASH diet, control of diabetes and obesity, and healthy life style have been recommended for acquired solitary kidney patients. |
Key Findings Related to Risk Factors, Outcomes, and Management of Solitary Kidneys
4. Conclusions
Solitary kidneys are of significant importance from a nephrologist’s perspective. Clinicians should be aware of the embryology, evolution, and potential complications of solitary kidneys over time. The fate and outcome of a solitary kidney are not always benign. The CSK may be associated with CAKUT, mesonephric duct (Wolffian duct)-derived structural anomalies in males, and Müllerian duct-derived structural anomalies in females. The CSKs have a higher number of nephrons and a greater GFR compared to acquired solitary kidneys. Initially, acquired solitary kidneys contain 50% of the nephrons, but the lower nephron number is partially compensated by hyperfiltration and hypertrophy later in life.
Individuals with CSKs with ipsilateral CAKUT and/or without compensatory enlargement may exhibit proteinuria, systemic hypertension, and a decrease in GFR. In contrast, individuals with acquired solitary kidneys may have favorable long-term outcomes but are at a higher risk of developing CKD and ESRD compared to those with CSKs. Avoiding the antenatal use of nephrotoxic drugs such as NSAIDs and aminoglycosides can help prevent the occurrence of CSK. The use of RAAS inhibitors is recommended after the age of one year to treat hypertension and proteinuria, improving outcomes for patients with CSKs. Lifestyle modifications, nutrition, and dietary management are recommended to enhance the longevity of individuals with solitary kidneys.
