




1. Introduction
Histiocytic sarcoma (HS) is a very rare non-Langerhans histiocytic disorder of unknown cause, with only a few hundred cases reported in the literature (1). HS may occur as an isolated disease or in the circumstances as other hematologic neoplasms, such as follicular lymphoma, myelodysplasia, or acute lymphoblastic leukemia (2). HS has been diagnosed in all age groups, especially in adults (2). There are no recognized environmental or hereditary genetic factors that lead to the development of HS (2).
2. Case Presentation
The patient is a 56-year-old man presented by a six-month history of a cutaneous plaque lesion on his left arm. The physical examination revealed a 10 × 15 cm violaceous erythematous indurated plaque with scaling on the posterior surface of the left arm without warmth and tenderness (Figure 1A).

A, Before treatment; B, after chemoradiotherapy
A biopsy of the skin lesion was performed. Histopathologic examination showed an infiltrating neoplasm was composed of highly atypical cells with the sarcomatoid features, hyperchromatic and reniform-appearing nuclei with prominent nucleoli and abundant eosinophilic cytoplasm as well as numerous mitotic figures, with some medium to large cells with convoluted nuclei and pale eosinophilic cytoplasm admixed with some small size lymphocytes, cellular debris, and nuclear dust (Figure 2). Neoplastic cells infiltrated the whole reticular dermis, without epidermotropism, extending to the deep layer of the dermis and dissecting collagen bundles, leading atrophy of pilosebaceous units.
; B, medium power shows large cells with hyperchromatic nuclei and eosinophilic cytoplasm admixed with some small lymphocytes (H & E, 20× magnification).")
A, Low power examination of histiocytic sarcoma involving entire reticular dermis without epidermotropism (H & E, 4× magnification); B, medium power shows large cells with hyperchromatic nuclei and eosinophilic cytoplasm admixed with some small lymphocytes (H & E, 20× magnification).
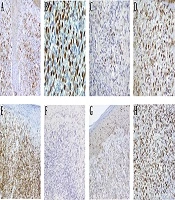
Immunohistochemistry revealed that the diffuse immunoreaction of tumoral cells for CD45 and MUM1, strongly positive for lysozyme and patchy immunoreaction for CD56, CD68, and S-100. Weak and focal Staining for S100, was seen, which suggested a degree of dendritic cell differentiation (3). The proliferation marker of Ki-67 revealed 90% immunoreactivity also, T cell reactivity of CD4 and CD5 were also evident. No immunoreaction for CD3, CD10, CD20, CD30, CD34, CD56, ALK1, CD1a, granzyme, BCL6, CD19, MPO, HMB-45, and cytokeratin (Pan-CK, CK7 and CK20) were identified. Ki-67 staining showed a high proliferative index of about 90% (Figure 3).

A, Immunohistochemistry staining shows positive immunoreaction for CD68; B, MUM1; C, lysozyme; D, S-100; E, LCA; F, and negative immunoreactivity for Granzyme; G, CD1a; and H, Ki-67 shows a high proliferative index of about 90%.
Laboratory evaluation, including complete blood count and comprehensive metabolic panel, were within the normal limits. To evaluate the extent of the disease, a PET (Positron emission tomography)-computed tomography (CT) scan was conducted, the result of which was normal except for increasing uptake in the posterior surface of the left arm.
The bone marrow biopsy results showed a normocellular bone marrow without abnormal changes, stromal injury, granulomas, or malignant neoplasm. The brain, chest, and abdominopelvic CT scan were normal. Since surgical resections were not considered as appropriate, the patient underwent aggressive multiagent chemotherapy with six cycles of ICE (ifosfamide, mesna, carboplatin, and etoposide) regimen and radiotherapy. After the chemoradiotherapy, the skin lesion completely disappeared (Figure 1B). The patient is currently visiting us for follow up without any recurrence of the disease.
3. Discussion
HS is a problematic and controversial group of uncommon hematopoietic neoplasms (4). Clinically, HS appears to show some special features, including frequent extranodal presentation, poor prognosis, and a possible association between tumor size and poor prognosis (4). The clinical course of HS is usually aggressive, and the presentation varies depending on the involved organ (5). Organ involvement may be asymptomatic and observed at the time of radiologic imaging (5). The patients who have unilateral or multichannel disease are more involved with the intestines, skin, and soft tissues, palpable mass lesions, and compressions of adjacent organs, such as intestinal obstruction, or constitutional symptoms (e.g., fever and weight loss) (6). Skin involvement has several potential symptoms ranging from a rash to tumorous swelling (4, 6). Laboratory abnormalities depend upon the affected organ system (4).
HS is diagnosed according to the pathologic evaluation of involved tissue interpreted within the clinical context (7). An excisional or incisional biopsy of affected tissue is preferred, over a fine needle aspirate or core needle biopsy (7).
Also, a localized form of de novo HS has a proper prognosis, but the disseminated disease typically has an aggressive clinical course and poor response to treatment (8). Histopathologic differential diagnosis includes Langerhans cell histiocytosis (LCH), Rosai-Dorfman disease (RDD), myeloid/monocytic sarcoma (MMS), diffuse large B cell lymphoma (DLBL) and anaplastic large cell lymphoma (ALCL), melanoma and carcinoma (3, 9). The absence of epidermotropism and intraepidermal cells and negative immunoreaction for CD1a are against LCH. No emperipolesis is noted in favor of RDD. The negative reaction for CD34 and MPO argue against the diagnosis of MMS. Negativity for CD20/BCL6 and CD30/ALK1 is against the diagnosis of DLBL and ALCL, respectively. Melanoma (S-100 and HMB-45) and carcinoma markers (Pan-CK, CK7, and CK20) show negative reactivity. Histomorphologic and immunostaining results are consistent with the histiocytic sarcoma.
To achieve the best management of patients with HS, the initial evaluation must confirm the histologic diagnosis, and extend the site of disease and the performance status of the patients’ Pretreatment evaluations in patients with the diagnosis of HS including laboratory tests such as CBC, serum electrolyte, and chemistries with liver and renal function, CT, or PET/CT to document organ involvement and BM biopsy for patients with cytopenias (4).
There is no standard treatment regimen for patients with HS. In patients with the unifocal disease, the protocol is surgical excision with adjuvant radiotherapy, depending on location and extends of disease (10). If radiotherapy is not available or very toxic, the postoperative observation is recommended (10). The protocol is aggressive multiagent chemotherapy with six cycles of ICE (ifosfamide, mesne, carboplatin, and etoposide) for patients with multifocal disease. Allogeneic hematopoietic cell transplantation should be considered for patients with relapsed or refractory disease who achieved remission with salvage therapy (7, 11).
