



1. Introduction
Atypical rhabdoid/teratoid tumor (ATRT) of the central nervous system (CNS) is an uncommon, fast-growing and malignant tumor of the brain and spinal cord, with incidence rate estimation of less than 1/1,000,000 (1). It mostly occurs in the cerebellum or the brainstem (2), often in children younger than 3 years of age (3), but it can also happen in older children and adults. AT/RT shares some of its morphological features with medulloblastoma, with close resemblance to choroid plexus papilloma (3). The histological hallmark of AT/RT are rhabdoid cells, which demonstrate large and eccentrically placed nuclei with a prominent nucleolus, in a brightly eosinophilic cytoplasm, with or without fibrillary globoid inclusions (3). Due to the rarity of the disease, there has been no consensus regarding the optimal treatment of this tumor (2). In this report, a case of AT/RT with rather unusual features was presented.
2. Case Presentation
A 17-year-old female with a sudden weakness of the left side of her body, along with painless urinary retention was brought to the emergency room. She gave a history of progressive low back pain for the past two months, but had no prior surgical history and or comorbidities.
The weakness of her left lower limb with strength limited to active movement only in the absence of gravity (score of 2 on the muscle power scale) was revealed by performing neurological examination. Perineal anesthesia was also detected. The results of other examinations showed normal limits for her limbs.
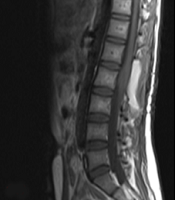
On magnetic resonance imaging (MRI) of the spine, intramedullary mass with abnormal signal in anterior thecal sac at L2 level was detected (Figure 1A and B).

Midline sagittal magnetic resonance imaging of the lumbar spine; A, preoperative T1- weighted; and B, T2-weighted imaging demonstrate an intramedullary mass with abnormal signal in anterior thecal sac, measuring 29 × 15 mm at the L2 level; C, postoperative T1-weighted; and D, T1-weighted with contrast imaging demonstrating residual tumor, with abnormal enhancement in left anterolateral aspect of posterior side of thecal sac and cord with extension to neural foramina and left neural canal at the L1/L2 level.
The patient underwent surgery, in which the intramedullary intradural tumor at the level of L2
was incompletely resected. Post-operative spinal MRI, along with contrast study suggested the presence of residual tumor (Figure 1C and D). The brain MRI obtained at this time was normal.
Histologic evaluation revealed a small blue round cell tumor (Figure 2A and B) which stained positive for actin, vimentin, and epithelial membrane antigen (EMA), cytokeratin (CK), CD99, and neuron specific enolase (NSE) after performing immunohistochemistry. However, the tumor stained negative for synaptophysin, chromogranin, and glial fibrillary acidic protein (GFAP) (Figure 2C). Due to Ki67 expression of 60% and loss of integrase interactor 1 (INI1) expression, a diagnosis of malignant embryonal tumor grade 4 (AT/RT, World Health Organization grade IV) was established. The diagnosis was confirmed by a pathology review, and positive staining for synaptophysin and negative staining for CD45, S100, and olig2 were documented by further IHC evaluations.
 showing proliferation of cells with scant, sometimes clear cytoplasm with high mitosis and high atypia and areas of necrosis characteristic of malignant small round cell tumor; C, positive staining for CD99, dot-like, paranuclear, and membranous pattern.")
Histologic features of tumor tissue; Hematoxylin and eosin staining (A, represents × 400 magnification; B, represents × 100 magnification) showing proliferation of cells with scant, sometimes clear cytoplasm with high mitosis and high atypia and areas of necrosis characteristic of malignant small round cell tumor; C, positive staining for CD99, dot-like, paranuclear, and membranous pattern.
Postoperatively, the patient underwent adjuvant radiotherapy, with neuroaxis dose of 3600 cGy, and localization to the tumor bed, to a total dose of 5220 cGy. She subsequently received a triweekly chemotherapy regimen of carboplatin and vincristine for eight cycles, without any significant side effects. After 20 months of follow-up, the patient was disease-free.
3. Discussion
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare medical condition occurring in the brain, with incidence estimated to be less than 1 per 1,000,000 (1), and is predominantly seen in the posterior fossa in early childhood (2). Its occurrence in the spine, especially during adulthood, is a rare presentation, and only a few cases have been reported (4). In cases of primary disease originating from the spine, it usually involves the cervical spine (5), whereas thecal sac involvement is only reported in a very advanced case, causing death a month after diagnosis (6). AT/RT lesions mainly occur intradurally, and intramedullary (7). Our patient presented with an intramedullary mass at the level of second lumbar vertebra in the anterior thecal sac. In contrast to the poor prognosis associated with the previous report of thecal sac involvement, our patient had exceeded the median survival associated with this diagnosis in general.
AT/RT was defined by WHO 2016 classification of tumors of the CNS as alterations of either INI1 protein (SMARCB1 gene) or, more rarely, BRG1 protein (SMARCA4 gene) (8). Histologic evaluation of our patient showed small blue round cell tumor that stained positive for actin, vimentin, and epithelial membrane antigen (EMA), CK, CD99, NSE, and synaptophysin along with loss of INI1 expression (Figure 2A-C), which was consistent with the findings from previous reports (3, 4, 9).
The prognosis of AT/RT has been reported to be poor, with median survival of 12 months in the
pediatric population (6). The mean overall survival among adults has been reported to be 12.5 - 15 months (9, 10), but there are reports of long-term survival in the literature (e.g., a case of ATRT of occipital lobe in a 24-year-old who survived with residual tumor for 5 years (11), and another patient was disease-free for 17 years following treatment (12). Our patient had also exceeded the median survival, being disease-free for 20 months.
The rarity of ATRT impedes the treatment choice which is mainly made based on retrospective studies, where multimodal treatment is mostly administered (9, 13). A case series from St. Jude Children Research Hospital, Tennessee documented higher survival rates in children treated with upfront RT and alkylating-based chemotherapy (14). Following their ACNS0333 protocol, Children’s Oncology Group documented improved survival by providing high dose chemotherapy and consolidative RT as well as using two cycles of induction chemotherapy of vincristine, methotrexate, etoposide, cyclophosphamide, and cisplatin, followed by three cycles of consolidation chemotherapy of carboplatin and thiotepa (15). Our case was treated after performing surgery, adjuvant RT, and alkylating-based chemotherapy, with a durable response to treatment.
3.1. Conclusions
Atypical teratoid/rhabdoid tumor is a rare and oftentimes aggressive tumor, which is mostly developed in the pediatric age group and in the posterior fossa. Our case, in line with several previous studies, produced encouraging results by combining modality treatments, including surgery, adjuvant chemotherapy, and radiotherapy.
