


1. Background
It has been more than 30 years since the first licensed recombinant therapeutic protein and insulin were introduced and commercially available (1). Recombinant protein production has completely changed human life and opened a new era. Nowadays, hundreds of therapeutic proteins from very different origins are produced in genetically modified organisms (1).
Interferons (IFNs) belong to a family of cytokines with antiviral, antiproliferative, and immunomodulatory properties (2). They are produced as a defense response to infections caused by viruses. IFNs are typically classified into 3 types based on their nucleotide sequence, interaction with specific receptors, chromosomal location, structure, and physicochemical properties. Type I IFNs are mainly composed of IFN-α and β, while IFN-γ is the sole member of type II IFNs. Type III IFNs comprise IFN-λ1, IFN-λ2, and IFN-λ3 (3). IFN-β (also called fibroblast IFN) is a 166-amino acid glycoprotein with a single glycan attached to the Asn80 residue. Its transcription is induced by viruses, endotoxins, mycoplasma, bacteria, and double-stranded polynucleotides (4, 5). Recombinant hIFN-β is considered a first-line treatment for relapsing-remitting multiple sclerosis (RRMS) (6, 7). Two kinds of recombinant hIFN-β have been produced for MS therapy. The non-glycosylated form of the recombinant hIFN-β (hIFN-β 1b) with 165 amino acids was first produced in Escherichia coli and approved by the US Food and Drug Administration (FDA) in 1993. In hIFN-β 1b, the Met1 residue was deleted, and Cys17 was replaced by a Ser to improve its bioactivity (8, 9). The glycosylated form of the recombinant hIFN-β (hIFN-β 1a) was then commercially produced in the Chinese hamster ovary (CHO) cell line. The problem associated with rhIFN-β produced in E. coli is its tendency to aggregate due to hydrophobic interactions (10). The carbohydrate moiety in the glycosylated recombinant hIFN-β covers the hydrophobic side chains, stabilizes protein folding, and decreases its aggregation (11).
Different mammalian expression systems have recently been used to produce glycosylated therapeutic proteins such as recombinant hIFN-β. Among those mammalian expression systems, the CHO cell line has attracted much attention in the commercial production of biotherapeutics (12). The CHO cell line confers several advantages over the other mammalian expression systems, such as the production of glycoproteins with glycan moieties similar to those found in mammalian glycoproteins, presence of powerful gene amplification systems, adaptability to growth in suspension and serum-free conditions, and low risk for the propagation of human viruses (13, 14). The long-term and large-scale production of recombinant proteins in mammalian cells depends on the stable integration of the transgene in the host genome. Multiple stable recombinant CHO cell lines have been constructed by a non-viral method for the large-scale production of recombinant proteins (15-17). In the current work, to generate a stable CHO-K1 cell line expressing the codon-optimized hIFN-β gene, the linearized codon-optimized human interferon-beta (opt-hIFN-β) sequence was transfected into CHO-K1, and stable genetically modified cells were selected based on the resistance to G418. The expression of the recombinant hIFN-β was confirmed by dot and western blotting for further scaling up in bioreactors.
2. Objectives
This study aimed to construct a stable CHO-K1 recombinant cell line expressing a codon-optimized recombinant hIFN-β gene. This is the first step toward the large-scale production of recombinant hIFN-β in bioreactors.
3. Methods
3.1. Construction of Recombinant Plasmid
The pOptiVEC vector harboring the opt-hIFN-β coding sequence was double digested with NotI and XbaI restriction enzymes and subjected to agarose gel electrophoresis to extract and purify the 621 bp fragment of opt-hIFN-β from the agarose gel using the Roche high pure PCR purification kit. The double digested opt-hIFN-β fragment was then cloned into the same sites (NotI and XbaI) in pcDNA 3.0 plasmid, downstream to the CMV promoter. The accuracy of the cloning steps and the opt-hIFN-β sequence were then verified using restriction enzyme digestion of the recombinant plasmid, polymerase chain reaction (PCR), and high-quality Sanger sequencing. The final recombinant expression plasmid was named pcDNA3-hIFN-β.
3.2. Maxi Preparation of Plasmid
Upon verification of error-free clones, the pcDNA3-hIFN-β recombinant plasmid was amplified in the DH5α strain of E. coli and purified using the Genopure Plasmid Midi Kit (Roche, Germany) according to the manufacturer procedure.
3.3. Cell Culture and Transient Transfection of CHO-K1 Cells
For transient transfection of CHO-K1 cells with the pCDNA3-hIFN-β recombinant plasmid, 24 hours prior to transfection, the cells were seeded in 24-well plates at a density of 2 × 105/mL cells in 500 µL DMEM/Ham’s F12 medium (50/50) media containing 10% FBS (Invitrogen, USA), 100 IU/mL penicillin, and 100 µg/mL streptomycin. The cells were incubated in a humidified incubator at 37°C and 5% CO2 to grow and reach approximately 60 - 70% confluence. At the time of transfection, the culture media were replaced with non-FBS and non-antibiotic DMEM/Ham’s F12 media. The cultured CHO-K1 cells were transfected with the circular pcDNA3-hIFN-β recombinant plasmid using lipofectamine 2000 according to the manufacturer’s instructions (Thermo Fisher Scientific, USA). In brief, 1 µL lipofectamine 2000 reagent was diluted in a 50-µL opti-MEM medium and incubated for 5 minutes at room temperature. About 1 µg of DNA was also diluted in a 50-µL opti-MEM medium, gently mixed with diluted lipofectamine 2000, and incubated for 20 minutes at room temperature. The mixture of DNA and lipofectamine 2000 was added to the CHO cells while swirling. Three to 4 hours after transfection, the culture medium was harvested, and a fresh, rich medium was added, containing 10% FBS (v/v), 100 IU/mL penicillin, and 100 μg/mL streptomycin. The cultured media were harvested every 24 hours after transfection for 3 days to verify protein expression using dot blotting analysis.
3.4. Stable Transfection of CHO-K1 Cell Line
For stable transfection of the CHO-K1 cells, the purified pcDNA3-hFIXβ plasmid was linearized using the PvuI restriction enzyme (Fermentas, USA) to facilitate its integration into the CHO-K1 genome. The linearized pcDNA3-hIFN-β plasmid was transfected into a CHO-K1 cell line using lipofectamine 2000 transfection reagent according to the manufacturer’s instructions. Twenty-four hours after transfection, the CHO-K1 cells were grown under the selective pressure of 450 µg/mL of G418 (Roche, Germany) for 1 month, during which the culture media were replaced with the fresh G418-containing media every 2 days. When the G418-resistant cells appeared, they were pooled and seeded in duplicates at a density of 3 × 105 cells in 2 mL of media per well of a 6-well plate. The supernatants were collected and subjected to enzyme-linked immunosorbent assay (ELISA) and dot and western blotting analyses.
3.5. Protein Expression Analyses
The supernatant of transiently transfected cells was collected every 24 hours for 3 days and subjected to dot blotting analysis. The culture media from stable-transfected CHO-K1 cells were collected and subjected to sandwich ELISA and dot and western blot analyses to detect the presence of recombinant hIFN-β in culture media.
3.6. Western Blotting
The supernatants from stably transfected cells were collected and subjected to SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) analysis. The SDS-PAGE analysis was performed according to the standard method described by laemmli in 1970 using 13% polyacrylamide resolving and 5% polyacrylamide stacking gels. The separated protein bands were electroblotted onto a polyvinylidene difluoride (PVDF) membrane and blocked in skim milk (5% w/v, in tris-buffered saline). The PVDF membrane was then incubated with mouse anti-hIFN-β monoclonal antibody (Millipore, USA), followed by incubation with HRP conjugated goat anti-mouse antibody (Millipore, USA). Immunoreactive bands were then visualized using Opti 4 CN substrate kit (Bio-Rad, USA).
3.7. Quantification of the hIFN-β Expression Using ELISA
Carbonate buffer containing 2 µg/mL polyclonal rabbit anti-human interferon beta was added to a 96-well plate (Millipore, USA) and incubated at 4°C for 16 hours. The plate was incubated in blocking buffer for 1 hour after disposing of coating materials. The mouse anti-human interferon-beta monoclonal antibody (DF 1:1000; Millipore, USA) as a detection antibody and goat anti-mouse HRP-conjugated antibody (1:5000; Millipore, USA) as a secondary antibody were added to the plate and incubated for 1 hour at room temperature. Then TMB substrate (Bio-Rad, USA) was used, and the absorbance was read at 450 nm wavelength using a plate reader (Thermo / LabSystems 352 Multiskan MS Microplate Reader, Artisan Technology Group, United States, Lab System 352).
4. Results
4.1. Construction of Recombinant pcDNA3-hIFN-β Plasmid Expressing opt-hIFN-β
The opt-hIFN-β coding sequence was digested and extracted from the pOptiVEC vector using NotI and XbaI restriction enzymes (Figure 1). The 621 bp fragment was then purified and inserted into the same places in the pCDNA 3.0 shuttle vector to construct recombinant expression plasmid pcDNA3-hIFN-β. To confirm the cloning steps, pcDNA3-hIFN-β recombinant plasmids were digested with NotI and XbaI and subjected to agarose gel electrophoresis to separate fragments related to the opt-hIFN-β coding sequence and linearized pcDNA3.0 vector. The accuracy of the recombinant plasmids was further verified by high-quality Sanger sequencing.

pOptiVEC digested with NotI and XbaI restriction enzymes. Lane 1, DNA ladder 1 kb; lane 2, pOptiVEC digested with NotI and XbaI; lane 3, uncut plasmid pOptiVEC.
4.2. Transfection of CHO-K1 Cells with pcDNA3-hIFN-β Recombinant Plasmid
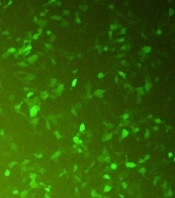
CHO-K1 cells were transfected with the recombinant pcDNA3-hIFN-β plasmid using the lipofectamine 2000 transfection reagent. During transfection, a green fluorescent protein (GFP)-expressing vector was used as a positive control to monitor the transfection efficiency indirectly (Figure 2). Untransfected cells were also used as a negative control. The transfection efficiency was estimated to be about 60 - 65% based on the green fluorescent signal from CHO-K1 cells transfected with the GFP-expressing vector monitored by fluorescence microscopy (Figure 2).

Transfected CHO-K1 cells with the GFP-expressing plasmid. The expression of GFP in the cytoplasm of the transfected CHO-K1 cell marks the cells green under fluorescence microscopy. The green cells represent the transfected cells with the GFP-expressing vector.
4.3. Expression of the hIFN-β in Transiently Transfected CHO-K1 Cells
To study the expression of recombinant hIFN-β by the opt-hIFN-β gene in transiently transfected CHO-K1 cells, 24, 48, and 48 hours after transfection, the culture media were harvested and subjected to dot blotting analysis, in which recombinant hIFN-β purified from E. coli was used as a positive control (Figure 3). As depicted in Figure 3, all the samples were positive in the case of hIFN-β expression in CHO-K1, so the study was continued to produce the stable CHO-K1 cell line expressing recombinant hIFN-β.

The dot blotting analysis of recombinant hIFN-β expressed by transiently transfected CHO-K1 cells. Supernatants were collected 24, 48, and 72 hours after transfection and subjected to dot blotting analysis using the mouse anti-hIFN-β primary antibody. The positive control was standard hIFN-β expressed by Escherichia coli.
4.4. Stable Expression of hIFN-β by CHO-K1
For stable expression, CHO-K1 cells were transfected with the PvuI-linearized pcDNA3-hIFN-β plasmid and grown under the selective pressure of 450 µg/mL of G418 for 1 month to develop stable CHO-K1 cells. The supernatant was collected from stably transfected cells and subjected to dot blotting analysis (Figure 4).
. Recombinant hIFN-β expressed by <i>Escherichia coli</i> was also used as a positive control (A).")
The dot blotting analysis of recombinant hIFN-β expressed by stably transfected CHO-K1 cells. Supernatants were collected from stably transfected CHO-K1 cells and subjected to dot blotting analysis using the mouse anti-hIFN-β primary antibody (B). Recombinant hIFN-β expressed by Escherichia coli was also used as a positive control (A).
4.5. SDS-Polyacrylamide Gel Electrophoresis and Western Blotting
The supernatant from stably transfected cells was harvested and subjected to SDS-PAGE, followed by western blotting analysis using the anti-hIFN-β monoclonal antibody. In this experiment, recombinant hIFN-β produced by E. coli and Cinnovex hIFN-β produced by CHO cells were used as positive controls (Figure 5 and lanes 1, 3, and 4). The band related to recombinant hIFN-β produced by stably transfected CHO-K1 cells appeared as heavy as Cinnovex hIFN-β and was heavier than recombinant hIFN-β produced in E. coli. The negative control was supernatant from non-transfected cells (Figure 5 and lane 5). As depicted in Figure 5, there was no band related to hIFN-β in non-transfected cells.

Western blot analysis was carried out on the supernatant collected from stably transfected CHO-K1. Supernatants from stably transfected and untransfected CHO-K1 cells were harvested and subjected to SDS-PAGE and western blot analysis using the anti-hIFN-β primary antibody. Lane 1, recombinant hIFN-β produced by Escherichia coli; lane 2, the Fermentase pre-stain protein ladder; lane 3, standard Cinnovex hIFN-β expressed by CHO cells; lane 4, the supernatant from stably transfected CHO-K1 cells; lane 5, supernatant from untransfected cells.
4.6. Quantification of the hIFN-β Produced by Stably Transfected CHO-K1 Cells Using Sandwich ELISA
To quantify recombinant hIFN-β produced by stably transfected CHO-K1 cells, the culture medium was harvested and subjected to sandwich ELISA. The exact protein quantity was estimated to be 126.13 ng/mL based on the standard curve.
5. Discussion
Two different protein expression systems are used for the production of recombinant hIFN-β for the treatment of MS. The non-glycosylated hIFN-β (IFN-β 1b) was first produced in E. coli, approved by FDA in 1993, and commercially available as BETASERON; hIFN-β 1a is produced in the CHO expression system with higher bioactivity (18).
Different prokaryotic and eukaryotic expression systems, including bacteria, yeasts, insect cells, filamentous fungi, and mammalian cell lines, are available for the production of recombinant proteins (19). Among them, mammalian cell lines are widely used to manufacture biotherapeutics due to their ability to produce a higher amount of secreted proteins in serum-free culture conditions and perform complex post-transcriptional modifications such as glycosylation. (20). About 60 to 70% of recombinant proteins are expressed in mammalian cells, primarily CHO cells, due to their ability to confer a glycosylation pattern close to that found in human cells (21). The CHO cell line has additional benefits, such as a safety profile in humans, accessibility of strong and diverse systems for gene amplification, and its adaptability to grow in suspension and serum-free culture conditions (21). However, compared to prokaryotic expression systems, eukaryotic systems have lower levels of protein production.
One strategy to improve heterologous protein production in mammalian cells is the codon optimization of the heterologous gene according to frequently used codons in host cells (22, 23). Previously, we designed a hIFN-β coding sequence according to the frequently used codons in CHO host cells in a way that the amino acid sequence remained unchanged. The optimized gene was cloned in the pCEP4 plasmid and transiently transfected into suspension-adapted CHO (CHO-s) cells. The expression efficiency of the hIFN-β protein from the optimized gene was compared to the unmodified sequence, indicating a 2.8-fold increase in the expression level of the hIFN-β by the codon-optimized gene (24). Based on our previous results, a pcDNA 3.0-based recombinant expression plasmid harboring the codon-optimized hIFN-β sequence (pcDNA3-hIFN-β) was constructed and transfected into CHO-K1 cell lines.
The expression of the recombinant hIFN-β was confirmed in transiently transfected CHO-K1 cells using dot blotting analysis. Then, the linearized pcDNA3-hIFN-β plasmid was transfected into CHO-K1 cells and grown under the selective pressure of 450 μg/mL geneticin for 30 days to develop stable cell lines. When the resistant colon appeared, they were pooled and seeded in a 6-well plate. The supernatant was collected 3 days later, and the expression of the recombinant hIFN-β was confirmed by western blotting and ELISA analyses. The western blotting analysis of the supernatant collected from stably transfected CHO-K1 cells showed a band with the same molecular weight as Cinnovex hIFN-β commercially produced by CHO cells. However, the band was heavier than the bacterial produced recombinant hIFN-β. The difference in molecular weight between prokaryotic and eukaryotic hIFN-β was expected and attributed to the occurrence of the N-glycosylation in the case of the recombinant hIFN-β produced in CHO (25). N-glycosylation is a co- or post-translational modification that occurs at the Asn-Xaa-Ser/Thr sequon on proteins produced in eukaryotes (26). The quantification of the recombinant hIFN-β production by stably transfected CHO-K1 cells was performed using ELISA and estimated to be 126.13 ng/mL. In this study, CHO-K1 stable cells producing recombinant hIFN-β were constructed. The next step is the isolation of stable cell lines with high productivity for scale-up in bioreactors, which can be a laborious and time-consuming process.
