

1. Introduction
Citrullinemia Type I (CTLN1) is a urea cycle disorder (Figure 1) caused by a deficiency in argininosuccinate synthetase (ASS), an enzyme that converts citrulline and aspartate into argininosuccinate. This condition can lead to hyperammonemia and encephalopathy in newborns or infants, often resulting in death or severe complications for those who survive. A milder phenotype of CTLN1, observed in late infancy or childhood, may allow for extended survival but is often associated with mental retardation (1, 2). In contrast, mutations in the SLC25A13 gene located on chromosome 7q21.3, which encodes the mitochondrial solute carrier protein citrin, are responsible for citrullinemia type 2 (CTLN2). Citrin deficiency impairs the shuttling of aspartate and glutamate to and from the mitochondrion, leading to mild hyperammonemia and a less pronounced phenotype. Clinical symptoms of CTLN2 typically appear later, and elevated plasma citrulline levels are less pronounced, with this type predominantly observed in East Asia. Based on clinical findings, it is possible to distinguish between the different types of citrullinemia (3).
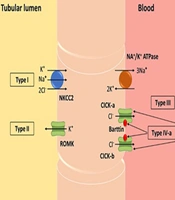
Bartter syndrome (BS), also known as congenital hypokalemic hypochloremic hypercalciuria with metabolic alkalosis, is characterized by excessive urinary excretion of sodium, chloride, and potassium. This disorder is classified into several subgroups, as shown schematically in Figure 2. Patients with BS typically present with hyperreninemic hyperaldosteronism and low to normal blood pressure values. Bartter syndrome can be divided into two distinct forms: Antenatal and classical BS. The antenatal form is more acute and associated with maternal polyhydramnios, while the classical form manifests its symptoms later and is milder (4).
")
Urea cycle (2)
 through their urine.")
A schematic illustration of Bartter syndrome, lose excess amounts of salt (sodium chloride) through their urine.
We describe a pediatric case of CTLN1 diagnosed at 5.5 years of age and treated with ammonia-reducing agents. The patient had a history of recurrent vomiting, gastroesophageal reflux disease, and kidney stones observed on ultrasound. She also presented with newly diagnosed BS, which was successfully treated with potassium and indomethacin. To our knowledge, this is the first case report describing a patient with CTLN1 accompanied by BS.
2. Case Presentation
We report on a 9-year-old Caucasian girl from a consanguineous marriage with a birth weight of 2.4 kg. The patient was born at a local hospital to a primigravida by vaginal delivery after a full-term, uncomplicated gestation. She had been generally well until 6 months of age, when her mother noticed that the baby had not yet begun rolling over from her stomach to her back and vice versa, reaching out for objects, or sitting without assistance. Symptoms of neurodevelopmental delay continued when the patient started walking at 19 months of age and began one-word speech at 24 months. She developed poor feeding, failure to thrive, and weekly recurrent projectile vomiting accompanied by night restlessness. The nonbilious nature of the emesis was consistent with gastroesophageal reflux disease, for which she was treated with omeprazole (1 mg/kg) for 15 months, and nutritional supplements including Carnitine, Biotin, and Zinc Plus were started. Celiac screening with serum tissue transglutaminase levels was normal. An abdominal ultrasound was obtained, showing no evidence of hypertrophic pyloric stenosis. Three months later, vomiting began to gradually fade away, while nighttime restlessness persisted. When she took ibuprofen on occasion, her condition improved markedly; however, due to concerns about long-term administration, the drug was discontinued.
Due to associated urinary symptoms, she was referred to a pediatric nephrologist at another center. Serial kidney sonography revealed multiple microlithiases (the largest being 6 millimeters) in the right kidney. She was started on Polycitra-K solution, hydrochlorothiazide, and Vitamin B6. Cystinuria and hyperparathyroidism were ruled out before hypercalciuria (urinary calcium/creatinine ratio of 0.55) and hyperoxaluria (31.6 milligrams per liter) were confirmed by spot urine analysis.
At 5.5 years of age, the patient experienced vomiting, low-grade fever, and several attacks of loss of consciousness and convulsive seizures. Encephalitis was diagnosed, and phenytoin was commenced. Due to the patient’s unexplained lethargy and somnolence, metabolic tests were performed, revealing an ammonia level of 100 µmol/L (normal range: 17-80). Hyperammonemia raised concern for a urea cycle disorder. Laboratory findings at this time were as follows: Serum sodium 140 mmol/L, potassium 4.8 mmol/L, calcium 8.4 mmol/L, arterial pH 7.47, pCO2 32 mmHg, and HCO3 27 mmol/L, with a base excess of +1.6 mEq/L, showing a mixed respiratory and metabolic alkalosis. Citrulline levels were 351 µmol/L (normal range: 10 - 45) and glutamine levels were 735 µmol/L (normal range: 345 - 685) in the HPLC plasma amino acid analysis, confirming the diagnosis of CTLN1. Ammonia-reducing agents (L-arginine, sodium phenylacetate, sodium benzoate, and carnitine) were initiated. Post-treatment, the parents reported continuous fever. Following a normal lumbar puncture, intravenous antibiotics were started empirically until the sepsis work-up results were remarkable for gram-negative pathogens. She was discharged from the hospital in good health.
Now, at 9 years old, the patient was admitted to the emergency department (ED) of our institution, a university hospital specialized in pediatric care, for severe dehydration secondary to a prolonged course of diarrhea and vomiting. There was a history of gastroenteritis and viral respiratory tract infections. On admission, the patient’s serum electrolyte levels were as follows: Potassium, 2.7 mmol per liter; calcium, 8.4 mg per deciliter; sodium, 138 mmol per liter; chloride, 109 mmol per liter. Intravenous potassium was started, and treatment was later changed to oral Slow-K and spironolactone, after which serum potassium returned to the normal range (4 mmol per liter). Hypokalemic hypercalciuria and metabolic alkalosis persisted for several days in the hospital. Serum magnesium was normal at 2.5. A 24-hour urine collection revealed the following: Sodium, 38 mmol per liter; chloride, 88 mmol per liter; potassium, 52 mmol per liter; and calcium, 70 mmol per liter. Ultrasound findings were notable for two stones, measuring 5 mm in the right kidney and 3 mm in the left kidney. On follow-up, she had to be hospitalized again 3 weeks later for repeated episodes of chronic diarrhea and vomiting. She was found to be severely hypokalemic with an increased transtubular potassium gradient (potassium 2.3 mmol per liter, TTKG 7.5). Once her acute hypokalemia was corrected with IV potassium resuscitation and Slow-K to 5.3 mmol per liter, the patient had normal blood pressures with no metabolic alkalosis (pH 7.35, HCO3 19.9 mmol per liter). Elevated plasma renin activity at 128.4 micro international units per milliliter (normal range: 2.99 - 35.4) and plasma aldosterone at 510.3 picograms per milliliter (normal range: 12 - 400), along with the patient’s background findings, were consistent with the diagnosis of BS. The patient was treated with indomethacin (0.4 milligram per kilogram) and Slow-K. Intermittent decreases in serum potassium were managed, and Polycitra-K solution was discontinued in the immediate follow-up period.
3. Discussion
To our knowledge, there are no previous case reports showing that urea cycle disorders are accompanied by salt-wasting tubulopathies. The clinical hallmark of our case included consistent vomiting from an early age until late childhood. More precisely, vomiting initially occurred alongside irritability and restlessness before progressing to chronic vomiting and metabolic alkalosis. Despite the uncommon association, CTLN1 and BS should be considered in the differential diagnosis of pediatric chronic vomiting after more common causes—such as allergic, endocrinologic, infectious, and structural causes—have been carefully excluded.
Bartter syndrome and CTLN1 share comparable clinical symptoms when the presentation is insidious. The insidious presentation of CTLN1 includes failure to thrive, behavioral abnormalities, and gastrointestinal symptoms (5). In a catabolic state, the patient may remain asymptomatic but is still at risk for developing an acute metabolic crisis. Failure to thrive and feeding difficulties were the first symptoms observed at 3 weeks of age. Although there is no definitive explanation for how hyperammonemia or hypochloremic metabolic alkalosis contribute to failure to thrive in this child, it is likely that earlier diagnosis of hyperammonemia could have prevented subsequent seizures.
Citrullinemia Type I is a devastating disorder that can significantly impact a child’s development; however, neurological prognosis depends on the duration of hyperammonemia (6). Our patient displayed symptoms of neurodevelopmental delay as early as 6 months of age, which were suggestive of hyperammonemia in CTLN1. Common neurologic presentations of CTLN1 include lethargy, respiratory distress, and coma, consistent with the respiratory alkalosis observed in our case in the initial blood sample taken at 5.5 years of age. However, other more challenging causes of chronic metabolic alkalosis should be considered in the setting of CTLN1. Persistence of metabolic alkalosis despite targeted treatment for hyperammonemia should prompt further investigation for a coexisting BS.
Frequent vomiting in BS can induce severe alkalosis due to chloride depletion, with serum bicarbonate levels potentially rising as high as 80 mmol/L. Additionally, a decreased glomerular filtration rate in BS patients with end-stage renal disease can worsen metabolic alkalosis by preventing adequate excretion of accumulated bicarbonate anions (7). Our patient’s serum creatinine level was within normal ranges. Marked metabolic alkalosis can be associated with neurological symptoms. This core finding underscores that high metabolic alkalosis due to underlying BS could exacerbate an already altered mental status in CTLN1. Therefore, it may be prudent to check the ABG and serum potassium profile to rule out BS in CTLN1 patients with recurrent vomiting.
Chronic vomiting, abuse of laxatives or diuretics, and salt-losing tubulopathies such as Gitelman and BSs are conditions that must be differentiated from decreased blood potassium levels associated with metabolic alkalosis, elevated levels of renin and aldosterone, and low to normal blood pressure (8).
When vomiting is chronic, as seen in bulimia or anorexia nervosa, hypokalemia, metabolic alkalosis, and increased levels of renin and aldosterone can occur due to chloride loss and volume contraction (9). In this case, diagnoses such as bulimia or anorexia nervosa were excluded due to the absence of signs typically associated with persistent vomiting, such as scarring or ulceration on the back of the hands or dental erosion, and the presence of high urinary chloride levels consistent with active renal salt loss. Laxative abuse was also quickly excluded for similar reasons.
Considering all the laboratory findings, diuretic abuse might be a potential explanation. However, our laboratory did not have access to a urine diuretic screen, and no outside medications were administered during the patient's stay. Despite this, her potassium levels remained very low while she was in the ward.
Thus, the main differential diagnosis was between Gitelman and BSs. Bartter syndrome seemed to be the diagnosis in our case for several reasons: The absence of hypomagnesemia, the presence of hypercalciuria, and diuretic insensitivity to thiazide administration (8).
The clinical diagnosis in our case, characterized by nephrolithiasis, hypokalemia, hypochloremia, normal serum magnesium, metabolic alkalosis, hypercalciuria, hyperreninemia, hyperaldosteronism, and normal blood pressure, was consistent with BS. Genetic testing for the type of BS was not available at our center. Initially, the case was approached as one of diarrhea; however, unlike typical diarrhea cases, our patient was voiding well with unchanged urine output. Therefore, severe dehydration associated with recurrent vomiting and diarrhea, but with stable or even increased urine output, could be another diagnostic clue for BS in such cases.
Bartter Syndrome is further characterized by isosthenuric polyuria with isosthenuria and excessive renal prostaglandin E2 production. Since non-steroidal anti-inflammatory drugs (NSAIDs) prevent the production of prostaglandin by blocking the enzymatic activity of cyclooxygenase (COX), a response to NSAIDs could determine whether recurrent vomiting with chronic metabolic alkalosis is possibly caused by BS, as seen in our case. Fluid losses in these patients pose a serious risk to their lives; therefore, indomethacin or other NSAIDs are routinely administered to alleviate diuresis and saluresis (10).
Bartter syndrome requires lifelong, high doses of potassium supplementation along with aldosterone antagonists such as spironolactone and/or potassium-sparing diuretics such as amiloride (300 mg/day for spironolactone and 40 mg/day for amiloride). Depending on the case, other antihypokalemic drugs such as ACE inhibitors, angiotensin receptor antagonists, or direct renin inhibitors may be needed. However, it should be noted that these additional treatments may further lower already low blood pressure levels in some cases (11).
In conclusion, CTLN1 may present with kidney and gastrointestinal manifestations and can be accompanied by BS, so renal tubular function should be monitored during follow-up (6). Both CTLN1 and BS tend to impede weight gain, and early diagnosis with appropriate treatment can lead to catch-up growth.
