



1. Introduction
Familial hypercholesterolemia (FH) is a genetic disorder of lipoprotein metabolism presented by highly elevated plasma cholesterol levels. Atherosclerosis due to FH manifests primarily in adulthood and detrimental cardiovascular consequences due to FH commence in childhood (1). The early treatment of risk factors for FH can reverse the atherosclerotic changes in the arterial system (2). The cholesterol residuum in external tissues for this defection causes clinical manifestations, including tendon xanthoma xanthelasma, and corneal arcus (3). FH represents the phenotypic manifestation of abnormal lipoprotein metabolism caused by a variety of genetic abnormalities. Mutations in the low-density lipoprotein receptor (LDLR) are the cause of single-gene FH. Over 1,500 mutations in this gene have been detected (4, 5) and these account for more than 80% of cases of monogenetic FH (6). In addition to LDLR, two other sets of autosomal dominant mutations play a key role in the pathogenesis of FH; the first one is a defective apo-B100 component (7-9) and the second one is a gain of function mutation affecting pro-protein convertase subtilisin/kexin 9 (PCSK9) (10). However, the rare autosomal recessive form of FH, called autosomal recessive hypercholesterolemia, has been described in a few families (11, 12). In the present study, we report a case of rare mutation discovered from the direct mutation screening of all exons in the LDLR gene in one Iranian patient with HF.
2. Case Presentation
The patient was a 41-year-old Arab man (2011) whose parents were cousins and died from Atherosclerosis. The cardiologist confirmed FH by the diagnostic criteria for this man in Imam Hospital of Ahvaz. The Simon Broom criteria were used for FH diagnosis (total cholesterol > 290 mg/dL (LDL-C > 190 mg/dL) in adults). In addition, the physical finding of the patient included tendon xanthomas over the elbow, Achilles tendon, and corneal arcus.
2.1. Material and Methods
Five mL of peripheral blood was collected in EDTA. DNA was extracted by the standard salting-out protocol. LDLR Gene-specific PCR primers were synthesized for all exons (a list of the primers used is available on request). The primers (Primer3 software, Gen Fanavaran, Iran) for exon 10 amplification were F: ‘5-ACCCTGCAGGATGACACAA-3’ and R: ‘5-GCCCACTAACCAGTTCCTGA-3’ that amplify 654 bp fragment.
The PCR program was conducted under the following conditions: 100 ng of genomic DNA, 200 μM of deoxyribonucleotide triphosphates (dNTPs), 1.5 mm of MgCl, 2.5 units of SuperTaq polymerase, and 25 pmol of each primer. The amplification was carried out in 25 μL volumes and 35 cycles: 90°C for 1 minute, 58°C
2.2. Results
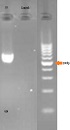
Gel electrophoresis for confirming PCR is illustrated in Figure 1.

The picture indicates exon 10 with 654 bp length and negative control for the same PCR
Direct sequencing analysis of the patient, after comparison with LDLR reference sequence (Gene ID: 3949), demonstrated a sporadic homozygous missense mutation GAC>AAC substitution in exon 10 of the LDLR gene in the patient (Figure 2) and heterozygous missense mutation in the siblings of the proband (Figure 3) (codon number: 492), which causes an amino acid exchange of aspartic acid to asparagine (Figure 2).
")
The result of genetic sequencing, indicating the homozygous missense mutation in exon 10 of the patient’s LDLR gene (sequencing with forward primer)
")
The result of genetic sequencing, indicating the heterozygous missense mutation in exon 10 of the proband’s siblings the LDLR gene (sequencing with forward primer)
3. Discussion
LDLR gene mutations are considered as an important factor causing FH. In 85% of patients, mutations concern the LDLR gene (13).
In a survey in Sweden, among 26 mutations that were detected in 50 subjects, 23 mutations were recognized in the LDLR gene. It was shown that almost 50% of patients have missense mutations, 27% have nonsense, and 12% have mutations in the splice site of the LDLR gene (14). A study in Albania reported two LDLR gene mutations accounting for FH (1646G>A, 81T>C). It was shown that nine patients were heterozygous for the 81T>C mutation in exon 2 of the LDLR gene, and 21 patients were heterozygous for the 1646G>A mutation (FH Genoa) in exon 11 (15). A Portuguese FH study represented that it is not possible to distinguish between patients with and without a mutation in the LDLR gene based only on lipid profile; thus, molecular diagnosis of familial hypercholesterolemia is essential to do. However, most of the diagnosed mutations were found in the LDLR gene (16).
A large number of methods have been used to screen for mutations in FH for research and clinical purposes, but direct sequencing is still considered the method of choice (17). We, therefore, performed a systematic screening of the coding region of the LDLR gene by the PCR-direct sequencing method. The present study is the first report of the LDLR variant in FH patients living in Khuzestan. We analyzed an affected person with FH using PCR and direct sequencing of coding exons of the LDLR gene.
As a result, we identified a genetic variant of LDLR in the FH patient. It was a missense mutation GAC>AAC located in exon 10 of the LDLR gene, which changes Aspartic acid to Asparagine. The LDLR gene encodes a protein with binding domains for apolipoproteins B and E (18, 19). Our found mutation changes the bioactivity of LDLR, resulting in FH; this finding can be useful for the detection of FH in Arab families.
