


1. Background
Toxigenic Vibrio cholerae is the causative agent of cholera, a severe dehydrating diarrheal illness, which is perceived as an emerging contagious infection with epidemic potential particularly in developing nations (1-3). Cholera transmission to humans happens through the ingestion of contaminated water or food. Aquatic environments serve as a supply for this bacterium (4). Cholera toxin (CT), which is encoded by the ctxAB genes, is the main virulence factor of V. cholerae (1, 5-7). Rapid, simple, and accurate analysis of V. cholerae isolates for the ability to produce CT provides essential benefits for the assessment of their clinical significance, as well as effective cholera surveillance system (8). The identification of toxigenic V. cholerae by culture, biochemical, and cholera toxin productivity test is time-consuming (three days) and laborious. Identification by PCR assays requires 4 - 5 hours for amplification, gel electrophoresis, and staining. Real-time PCR assays need the use of an extravagant instrument with experienced technicians (9), which routinely does not suit remote and resource-limited laboratories, as well as field application. As of late, a technique named Loop-mediated isothermal amplification (LAMP) technique has emerged as a simple rapid diagnostic tool for the detection of several pathogens. LAMP employs auto-cycling strand displacement DNA synthesis that is performed by Bst DNA polymerase large fragment without the need for thermal cycling (10). The LAMP assay has few advantages over PCR: (i) the whole procedure can be completed by placing all the reagents in a single tube incubated under isothermal conditions (60 - 65°C), (ii) utilization of four specially designed primers recognizing six discrete regions on the target nucleotides gives higher specificity (11), and (iii) agarose gel electrophoresis is not required because a positive reaction shows turbidity as a result of insoluble magnesium pyrophosphate by-product (12, 13).
2. Objectives
The aim of the present study was to develop a sensitive and specific real-time LAMP assay for simple and rapid quantification of toxigenic V. cholerae in the reaction.
3. Methods
3.1. Bacterial Strains and Genomic DNA Extraction
Overall 13 bacterial strains were used in this study, as follows: V. cholerae (toxigenic) ATCC14035; V. cholerae (nontoxigenic) CIP 104154; V. fischeri ATCC 7744; V. parahaemolyticus ATCC 17802; Yersinia enterocolitica ATCC 35669; Yersinia pseudotuberculosis ATCC 29833; Proteus vulgaris PTCC 1079; Pseudomonas aeruginosa ATCC 27853; Escherichia coli O157H7 ATCC 43895; Salmonella Typhi ATCC 700931; Shigella sonnei ATCC 9290; Staphylococcus aureus ATCC 25923; and Streptococcus pneumoniae ATCC 700669. Bacterial DNA was extracted from a single loopful of each bacterial culture using the QIAamp DNA Mini Kit (Qiagen) following the manufacturer’s protocol.
3.2. Primer Design, PCR, and LAMP Reactions
Four LAMP primers, VF3: TTTTCGTATACAGAATCTCTAGC; VB3: GCGTTTTATTATTCCATACACAT; VFIP: GGAAAAAGAGAGATGGCT; and VBIP: TTTTCGACTTTAGCTTCAGT, were designed based on the cholera toxin subunit B (ctxB) gene sequence (accession number: AY804244) using PrimerExplorer V. 4.0 software (http://primerexplorer.jp/e/). Primer specificity was confirmed by NCBI BLAST (www.ncbi.nlm.nih.gov/).
Conventional PCR using outer primers (VF3 and VB3) was carried out with an initial denaturation at 95°C for 3 minutes, followed by 30 cycles of 95°C for 10 seconds, 55°C for 10 seconds, and 72°C for 30 seconds, and a final extension at 72 °C for 2 minutes to amplify a 182-bp ctxB amplicon.
The LAMP reaction was performed in a final volume of 25 µL containing 1.6 µM of each of FIP and BIP primers, 0.2 µM of each of F3 and B3 primers, 1.4 mM of each of the dNTPs, 0.8 M betaine (Sigma, USA), 6 mM MgSO4, 8 U of the Bst DNA polymerase large fragment (New England Biolabs, USA), and 2 µL of the template DNA. Likewise, calcein reagent (25 µM) (Dojindo, Japan), as a fluorescent metal indicator, was added to the reaction tubes to facilitate the visual inspection of positive LAMP reactions under a UV lamp. The LAMP reaction mixture was incubated at 65°C for 1 hour, followed by enzyme inactivation at 80°C for 5 minutes according to the LAMP protocol (14). No template DNA control was included in each run of the LAMP reaction.
The intensity of turbidity in each LAMP reaction tube was graphed using a real-time turbidimeter (LA-320; Eiken, Japan). A positive reaction was defined as a reaction with turbidity reaching the threshold value (0.1) within the 60-minute incubation time according to the LA-320 software package.
To verify the turbidity and color change observations, the LAMP reactions were additionally examined by electrophoresis on the 1.5% agarose gel.
3.3. TA Cloning and Preparation of Standard Plasmid
A fragment of 260 bp from the ctxB gene was amplified by primers CT-F: CACAAATACATACGCTAAATG and CT-R: CATACTAATTGCGGCAATC. The PCR product was then purified with the PCR Purification Kit (Bioneer). The purified ctxB gene fragment was ligated into the pTZ57R/T vector using 1U of T4 DNA ligase following the manufacturer’s protocol of InsTAcloneTM PCR Cloning Kit (Fermentas), followed by the transformation of E. coli JM107 competent cells and blue/white screening for the identification of recombinant clones. Subsequently, the plasmids of the selected white clones were extracted by the AccuPrep Plasmid Mini Extraction Kit (Bioneer) and the recombinant plasmids containing ctxB were then confirmed by PCR using outer primers (VF3 and VB3). The recombinant plasmid, designated as pTZ-ctxB, was used to make dilutions as a reference in sensitivity testing and quantitative evaluation and positive control in the LAMP assay.
3.4. Sensitivity of LAMP Assay
To evaluate the detection limit of the LAMP in comparison with PCR, the pTZ-ctxB plasmid created using the aforementioned cloning was adjusted to the concentration of 2.8 pg/μL, and diluted into 10-fold series (8.3 × 105 to 8.3 × 10-4 copies) per reaction tube. The use of the plasmid DNA standard in the quantification of either LAMP or PCR is more convenient than the use of genomic DNA.
Gene amplification in a LAMP reaction tube was detected using a real-time turbidimeter (for real-time turbidity measurement) and visual inspection by the naked eye either as turbidity or in the form of green fluorescent of calcein under a UV lamp.
The detection of PCR products and the verification of LAMP results were performed by electrophoresis on the 1.5% agarose gel. To quantify the ctxB gene copy number using the real-time turbidimeter, a standard curve was constructed by plotting the Tt (Time threshold) values against the log copy number of pTZ-ctxB plasmid standard. Then, linear regression was computed using the Microsoft Excel program.
3.5. Determination of LAMP and PCR Specificity
The specificity of LAMP assay versus PCR was examined using extracted genomic DNA (50 ng) from a set of aforementioned Gram-negative and Gram-positive bacterial strains. The LAMP reactions were monitored with a real-time turbidimeter. Electrophoresis on the 1.5% agarose gel was carried out to detect the PCR products and to verify the LAMP results.
4. Results
4.1. Sensitivity of LAMP Assay
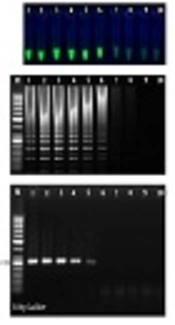
The sensitivity of the LAMP and PCR assays was determined using 10-fold serial dilutions of the pTZ-ctxB plasmid standard of a known copy number of the ctxB gene. After LAMP and PCR, 5 µL of each of the amplified products was subjected to electrophoresis on the 1.5% gel. The limit of detection for our ctxB-based LAMP assay using either the visual inspection of green fluorescent of calcein under a UV lamp (Figure 1A) or the electrophoretic detection of the amplified products (Figure 1B) was determined to be 8.3 copies of the ctxB gene per reaction while ctxB-based PCR required at least 83 copies of the ctxB gene per reaction for having a visible band on the agarose gel (Figure 1C). Therefore, the LAMP amplification technique was more sensitive than the PCR technique. This finding is in agreement with earlier studies comparing the sensitivity between LAMP and PCR (15, 16).
; B, electrophoretic analysis of <i>ctxB</i>-based LAMP reactions. The ladder-like pattern on the agarose gel (1.5%) indicates positivity; C, electrophoretic analysis of <i>ctxB</i>-based conventional PCR products; Lane M, 50-bp DNA ladder. (#1 - 10: 2.8 to 2.8 × 10<sup>-9</sup> pg/μL of pTZ-<i>ctxB</i> plasmid as the template).")
Sensitivity testing of ctxB-based LAMP and PCR assays; A, positive LAMP reactions fluoresced green under UV (tubes 1 - 6); B, electrophoretic analysis of ctxB-based LAMP reactions. The ladder-like pattern on the agarose gel (1.5%) indicates positivity; C, electrophoretic analysis of ctxB-based conventional PCR products; Lane M, 50-bp DNA ladder. (#1 - 10: 2.8 to 2.8 × 10-9 pg/μL of pTZ-ctxB plasmid as the template).
4.2. Quantification of ctxB Gene Copy Number by Real-Time LAMP
Benefiting from the exquisite sensitivity of LAMP assay, the quantification of ctxB gene copy number of V. cholerae was accomplished with the help of a real-time turbidimeter via the generation of a standard curve. Serial dilutions of the plasmid DNA (with known ctxB gene copy number) were plotted against the time of positivity (time threshold, Tt) in LAMP reactions (Figures 2A and 2B). Noteworthy is the consistency between real-time turbidimeter results (Figure 2A) and the observation of green fluorescent of calcein under a UV lamp (Figure 1A) and characteristic ladder-like multiple bands on the agarose gel (Figure 1B). Moreover, as seen in the graph (Figure 2B), a strong inverse linear correlation (r = 0.9, P < 0.001) was observed between the ctxB gene copy number versus the time of positivity in LAMP reaction tubes. Therefore, the quantification of unknown gene copy number in the LAMP reaction can be extrapolated from the standard curve on the basis of the known time of positivity.
; B, standard curve for the quantitative determination of toxigenic <i>V. cholerae</i> in water samples.")
Real-time turbidity measurements of LAMP reactions; A, representative turbidity graph for decreasing concentrations of the pTZ-ctxB plasmid (#1 - 6: 2.8 to 2.8 × 10-5 pg/μL corresponding to 8.3 × 105 to 8.3 copies/reaction of the plasmid); B, standard curve for the quantitative determination of toxigenic V. cholerae in water samples.
4.3. Specificity of LAMP Assay
The specificity of our LAMP assay was evaluated with extracted genomic DNA (50 ng per reaction) of 12 bacterial strains closely related to toxigenic V. cholerae. The genomic DNA of toxigenic V. cholerae merely showed a positive LAMP result based on the real-time turbidimeter results and the characteristic ladder-like multiple bands on the 1.5% agarose gel (data not shown). These results indicate that the assay was specific for toxigenic V. cholerae. A similar result was obtained by PCR, clearly indicating that the LAMP assay is as specific as PCR amplification.
5. Discussion
The frequent outbreaks caused by cholera toxin-producing V. cholerae in developing countries emphasize the need for the rapid and accurate identification of this organism. The bacterial culture test for the isolation and identification of toxigenic V. cholerae from environmental water samples needs plating the water sample onto selective agars, subculture, and cholera toxin productivity test, which takes 3 to 4 days. The PCR assay requires 4 - 5 hours of amplification, electrophoresis, and staining. Conversely, the LAMP assay is faster (less than 1 hour) and easier without the need for any cumbersome post-PCR analysis like agarose gel electrophoresis.
The performance of LAMP and PCR-based detection assays for V. cholerae has been compared in several reports. LAMP has been found with higher sensitivity than PCR yet with similar specificity (17, 18). Consistently, our data support the view obtained from earlier studies. While the detection of toxigenic V. cholerae by the LAMP technique based on the ctxA gene was previously reported by Yamazaki et al. (19) and Okada et al. (1), our real-time LAMP assay targets the ctxB gene of the toxigenic V. cholerae genome with the ability for the quantification of gene copy number in the LAMP reaction.
Our real-time LAMP assay could detect 8.3 copies of the ctxB gene in a reaction tube, which indicates its high sensitivity comparable with real-time PCR sensitivity for the detection of trace amounts of DNA in a sample.
In our study, a strong linear correlation (r = 0.9) between the ctxB gene copy number and the time of positivity indicates that the data obtained from the LAMP reaction were measurable. Therefore, the developed LAMP assay can be employed in the monitoring of the toxigenic V. cholerae DNA load in samples.
5.1. Conclusions
The LAMP method is preferred over conventional PCR as it is 3 - 4 times quicker, 3 - 6 times cheaper, and without the need for specialized equipment such as a thermal cycler or skilled personnel. In addition, the LAMP assay for the quantification of toxigenic V. cholerae DNA was highly sensitive and specific. Therefore, the developed LAMP assay could be a potential tool to monitor the presence and load of toxigenic V. cholerae DNA within environmental water samples upon further validation. This is especially useful in epidemiological studies and in resource-limited settings in developing countries for improving the management of cholera outbreaks.
