



1. Context
Liver cirrhosis, a severe consequence of chronic liver diseases, substantially disrupts immune homeostasis, predisposing patients to secondary infections (1). These infections are not just complications but also major contributors to the high mortality rates observed in cirrhotic patients (2). This review examines the immunological mechanisms underlying this vulnerability, emphasizing innate and adaptive immune disruptions, including Kupffer cell dysfunction, complement deficiencies, and T-cell abnormalities. Understanding these pathways is critical for developing targeted therapies to manage and prevent infections, improving outcomes for patients with hepatic cirrhosis (3).
2. Relationships Between Liver Cirrhosis and Infection
2.1. End-Stage Liver Disease and Liver Cirrhosis
The concept of end-stage liver disease (ESLD) was initially proposed in the 1980s, but a standardized definition emerged only in 2018 (2). End-stage liver disease encompasses advanced hepatic insufficiency caused by chronic injuries or conditions, such as acute-on-chronic liver failure (ACLF), decompensated cirrhosis, and hepatocellular carcinoma (4). This definition combines morphological and functional aspects of the liver, emphasizing that ESLD represents the terminal stage resulting from cumulative chronic liver injuries. It is characterized by progressive deterioration of liver function and progression to decompensation. Among patients with ESLD, liver cirrhosis is a common condition, representing a progressive liver disease caused by one or more etiologies, which can be divided into a compensated stage and a decompensated stage (5). Globally, cirrhosis-related mortality rose by 47.15% from 1990 to 2017, with a 12.02% increase in China (6). During the compensated stage, patients with liver cirrhosis often exhibit no obvious clinical symptoms and maintain a relatively good quality of life. However, as the disease progresses to the ESLD stage, clinical manifestations such as hypersplenism, gastrointestinal bleeding, ascites, jaundice, sepsis, and hepatic encephalopathy may emerge (7). Liver cirrhosis is often complicated by infections, which accelerate the clinical progression of the disease and lead to complications such as acute kidney injury, organ failure, and ACLF. Infection is not only a common complication of liver cirrhosis but also a critical factor that exacerbates disease progression, leading to complications such as acute kidney injury and organ failure (8).
2.2. Secondary Infections
In patients with liver cirrhosis, the incidence of bacterial infections is obviously greater than that in the general population. Medical studies have shown that the incidence of bacterial infections in hospitalized cirrhotic patients ranges from 25% to 40%. This is 4 to 5 times higher than the incidence in the general population. Furthermore, among patients with decompensated liver cirrhosis, the risk of death due to infectious complications is as high as 3.75 times greater (9, 10). These data highlight the importance of early diagnosis and effective management of patients with liver cirrhosis to reduce the risk of complications and death caused by bacterial infections. In abdominal infections, predominant pathogens include Escherichia coli, Klebsiella pneumoniae, and Staphylococcus aureus. Among abdominal infections, Escherichia coli is the most frequently encountered, followed by Klebsiella pneumoniae, Staphylococcus aureus, Enterococcus faecium, and Enterococcus faecalis (11). In respiratory infections, conditioned pathogens such as Pseudomonas aeruginosa and Staphylococcus aureus are more common. Notably, prolonged antimicrobial use in decompensated cirrhosis promotes drug-resistant nosocomial infections, increasing sepsis risk (12). Recent studies have further highlighted the seriousness of secondary infections in liver disease patients, as these infections can accelerate the progression of liver cirrhosis, especially in the decompensated stage, where patients are more susceptible to bacterial infections (2). Spontaneous bacterial peritonitis (SBP), one of the most frequent infections in cirrhosis, is primarily caused by Enterobacteriaceae and is associated with rapid progression to sepsis (13). Invasive fungal infections (IFIs), such as candidiasis and aspergillosis, although less prevalent, are particularly fatal due to delayed diagnosis and resistance to empirical antibiotics. In various medical studies, invasive IFIs are recognized as important causes of illness and death in patients with decompensated liver cirrhosis (14). Among all IFIs, Candida infections have the highest incidence rate, followed by Aspergillus infections (15). Given the unclear manifestation of infection and the complexity of diagnosis, rapid confirmation is often difficult, leading to delayed treatment and increased mortality risk. Therefore, early recognition and treatment of IFIs are vital for patients. To reduce mortality rates and improve the quality of life for this population, enhancing the understanding of these infections, improving diagnostic techniques, and developing effective prevention and treatment strategies are essential.
3. Immune Mechanisms and Susceptibility in Patients with Cirrhosis
Structural alterations in the liver that contribute to cirrhosis development include persistent hepatocellular injury, inflammation, and fibrotic remodeling (16). The interaction between the immune system and liver function is particularly intricate. Patients with cirrhosis often show susceptibility and adverse prognoses following sepsis due to disruptions in immune regulatory mechanisms (17, 18). Inflammatory reactions driven by the immune system have a crucial impact on the development of cirrhosis, and the coexistence of cirrhosis and elevated portal pressure exacerbates immune cell dysfunction and injury (19). The immunodeficient state in patients with decompensated cirrhosis involves abnormalities in various immune cells and factors, including IL-2, Kupffer cells (hepatic macrophages), monocytes and neutrophils, and the complement system (20). These abnormalities weaken the resistance to secondary bacterial infections in cirrhotic patients, escalating the risk of complications. Cirrhotic patients often suffer from various functional abnormalities of innate and adaptive immunity, which lead to a decrease in their resistance to secondary bacterial infections and an increased risk of complications (21). See Figure 1 for details.

Abnormal immune function increases the risk of infection in liver cirrhosis patients
3.1. Inherent Immune Abnormalities
3.1.1. Kupffer Cell Dysfunction
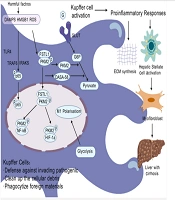
Immune cells, especially Kupffer cells and recruited macrophages, are key regulators of liver inflammation and play critical roles in the progression or regression of liver fibrosis. Upon hepatocyte injury, danger-associated molecular patterns (DAMPs) are released, which further activate Kupffer cells and recruit infiltrating macrophages (22). Once activated, macrophages release many cytokines that can directly damage the hepatic parenchyma, thereby impacting the overall health of the liver. Hepatic macrophages account for 90% of the total macrophages in the human body and exhibit significant heterogeneity, including resident macrophages and monocyte-derived macrophages (MDMs) (23). Kupffer cells, the resident macrophages of the liver, are predominantly distributed within hepatic sinusoids. Originating from yolk sac-derived progenitor cells, they colonize liver tissue during embryonic development and are also replenished through differentiation from bone marrow-derived monocytes. As the primary macrophages in the liver, Kupffer cells maintain hepatic homeostasis through self-renewal, phagocytosis of pathogens and cellular debris, and regulation of iron metabolism (24). Under normal conditions, Kupffer cells efficiently phagocytose and clear pathogens, bacteria, toxins, and other harmful substances from the bloodstream or intestine, maintaining hepatic health (25). The interaction between multiple cell types and molecules can cause liver cirrhosis, with macrophages, as important members of the immune system, playing a central role in this process. Hepatic macrophages promote liver fibrosis by increasing the survival rate of activated hepatic stellate cells (HSCs) in an NF-κB-dependent manner (26). Liver fibrosis is primarily driven by the activation of HSCs, a process triggered by persistent pathological stimuli such as chronic inflammation or metabolic stress. Nonparenchymal cells are activated, leading to abnormal expression of fibrillar proteins and related cytokines and triggering the proliferation or decomposition imbalance of fibrous tissue, resulting in excessive deposition of fibrous structures and the development of liver fibrosis, which may eventually progress to liver cirrhosis or liver cancer (27, 28). Furthermore, macrophage-derived transforming growth factor-β (TGF-β) has been identified as a key molecule that initiates HSC activation. Some studies have shown that Toll-like receptor (TLR-4 and TLR-9) signaling pathways mediate crosstalk between inflammatory and fibrogenic pathways (29). Under diseased conditions, the liver primarily relies on bone marrow-derived macrophages, which are recruited to the liver after the activation of HSCs and Kupffer cells and become important sources of replenishment and regeneration after hepatic macrophage depletion. The critical step in liver fibrosis is the activation of HSCs, which transform into myofibroblasts after hepatocyte injury, becoming the primary cellular source of fibrosis (30). Most studies indicate that under the combined action of various pathogenic factors, Kupffer cells in the liver are activated and promote HSC activation and extensive extracellular matrix synthesis under the influence of multiple pathogenic factors and external chemical mediators (31, 32). The pathways involved in HSC activation are complex and diverse and can be roughly divided into intracellular and extracellular sources. Various cellular signaling pathways can activate HSCs, such as nuclear receptors, G protein-coupled receptors, cell proliferation and fibrosis pathways, innate immune signaling pathways, adipocytokines and cytokines, and genetically related signal transduction pathways (33). Additionally, extracellular stimuli can promote HSC activation by secreting cytokines or activating signaling pathways (34, 35). In normal livers, Kupffer cells, as sentinel cells, dominate and maintain hepatic homeostasis. However, under pathological conditions, these cells undergo phenotypic changes, secrete anti-inflammatory or proinflammatory factors, and recruit more macrophages, namely, BMDMs, to the liver. These BMDMs are similar to Kupffer cells in terms of function and plasticity and have a crucial impact on the development and resolution of liver diseases (35). Recent studies on intracellular functional reprogramming have demonstrated marked upregulation of follistatin-like protein 1 (FSTL1) in fibrotic liver macrophages. These macrophages inhibit proinflammatory M1 polarization and NF-κB pathway activation both in vivo and in vitro (29). Follistatin-like protein 1directly binds to pyruvate kinase M2 (PKM2) through its FK domain, a critical interaction that promotes PKM2 phosphorylation and nuclear translocation. This binding mechanism not only reduces ubiquitination of PKM2 but also enhances glycolytic activity, ultimately leading to increased PKM2-dependent glycolysis and subsequent M1 polarization. Of particular significance, PKM2 serves as a key mediator of aerobic glycolysis - a metabolic process strongly associated with oncogenesis and inflammatory pathways (36). Recent evidence further demonstrates that PKM2 governs metabolic reprogramming in macrophages during inflammatory responses. Through its interaction with hypoxia-inducible factor 1α (HIF-1α), this enzyme activates HIF-1α-dependent transcriptional programs that are indispensable for sustaining aerobic glycolysis in macrophage populations. Furthermore, pharmacological activation of PKM2 (DASA-58) can alleviate FSTL1-regulated glycolysis and inflammation to a certain extent (37). Collectively, this study revealed that macrophage FSTL1 promotes liver fibrosis progression through intracellular PKM2 reprogramming in macrophages, inducing M1 polarization and inflammation (29). See Figure 2 for details.

Kupffer cell dysfunction leading to liver cirrhosis
In cirrhosis, impaired liver function and altered hemodynamics lead to a notable decline in both the number and activity of Kupffer cells, compromising pathogen clearance (38). Additionally, Kupffer cell activity is partially dependent on the activity level of plasma fibronectin. As liver function decreases, plasma fibronectin activity decreases, further impairing Kupffer cell function and allowing the accumulation of gut-derived bacteria and endotoxins in the body, increasing the risk of infection. From a clinical standpoint, this dysfunction highlights the potential benefit of therapeutic approaches aimed at restoring Kupffer cell function or targeting gut-derived endotoxemia, such as probiotics, rifaximin, or fecal microbiota transplantation in cirrhotic patients.
3.1.2. Defects in the Complement System
The liver plays a crucial role in the immune defense of the body, and the complement system, an essential component of the innate immune system, performs a critical function when liver function is severely impaired. The complement system participates in immune responses, in turn enhancing the body's ability to clear pathogens. However, in liver cirrhosis, impaired hepatocyte function disrupts the synthesis and regulation of complement proteins, resulting in deficiencies in complement activity and synthesis. Specifically, a reduction in crucial complement factors directly affects the normal function of serum opsonization, increasing the susceptibility of cirrhotic patients to bacterial infections. Susceptibility to infections in cirrhotic patients is intimately linked to disruptions in immune mechanisms. Factors such as elevated IL-2 levels, functional defects in Kupffer cells, abnormal immune function, and defects in the complement system, all stemming from liver cirrhosis, collectively contribute to a marked reduction in these patients' resistance to bacterial infections. Furthermore, the abnormal activation of immune-mediated inflammatory responses exacerbates the risk of infection, leading to poor patient outcomes.
3.2. Adaptive Immune Abnormalities
3.2.1. Impairment of T Follicular Helper Cells
T follicular helper (Tfh) cells, a specialized subset of CD4+ T cells, are essential for maintaining optimal immune function. These cells express the CXCR5 receptor, PD-1 molecules, and ICOS protein, all of which facilitate their interaction with B cells. Additionally, Tfh cells produce the cytokine IL-21 and regulate transcription factors such as C-MAF and BATF. The development of these cells is influenced primarily by the transcription factor BCL-6, with in vivo studies indicating that STAT5 plays a pivotal role in T-cell development and exerts an inhibitory regulatory effect on the generation and function of CD4+ Tfh cells (17). Specifically, when STAT5 levels are insufficient, the expression of Blimp1 is altered, leading to increased expression of Tfh cell-specific genes, such as BCL-6, MEF factors, BATF, and IL-21. IL-21 and BCL-6 regulate Tfh cell differentiation and maturation. IL-21 promotes Tfh cell differentiation and enhances the expression of surface CXCR5, while BCL-6 is critical for the migration of Tfh cells to lymphoid follicles and the formation of B-cell germinal centers (39). Refer to Figure 3 for a visual illustration of this mechanism.
 cells")
Mechanism by which IL-2 elevation damages T follicular helper (Tfh) cells
In patients with cirrhosis complicated by chronic endotoxemia, dysregulation of immune responses is evident, characterized by elevated levels of both proinflammatory and anti-inflammatory cytokines in the serum (40). A significant increase in the serum IL-2 level impairs the differentiation of Tfh cells by upregulating the transcription factor Blimp-1 and suppressing TCF-1 expression (41), ultimately compromising immune function. Once Tfh cells are impaired, the maturation of central B cells and the production of high-affinity antibodies are affected, leading to reduced humoral immune efficiency and an increased risk of infection. Nevertheless, studies supporting this mechanism are limited by small sample sizes and reliance on animal models, underscoring the need for broader clinical trials to validate these findings in human populations. Therefore, Tfh cells play a vital role in adaptive immunity, not only by promoting the proliferation of B cells and immunoglobulin class switching to stimulate the formation of germinal centers, which drives the differentiation of B cells into plasma cells and memory B cells but also by secreting IL-21 to enhance CD8+ T-cell function, thus contributing to cellular immunity.
3.2.2. Aberrant Expression of HLA-DR Molecules
Immunodeficiency in cirrhosis is caused by multiple factors, including the persistent stimulation of microbial- and damage-associated molecular patterns (MAMPs and DAMPs), reduced synthesis of nutritional factors by the liver, splenomegaly with immune cell sequestration in the spleen, and the underlying etiology of cirrhosis (e.g., alcohol or viral infection). This immunodeficiency culminates in “immune paralysis”, thereby elevating the risk of bacterial infections (42). The term “immune paralysis” is defined by the downregulation of HLA-DR expression on monocytes, and it is typically associated with immune dysregulation and a greater incidence of bacterial complications. In patients with decompensated cirrhosis, peripheral blood monocytes spontaneously produce a variety of proinflammatory cytokines, whereas monocytes from healthy individuals secrete only limited amounts of IL-8 and IL-6. These findings indicate a proinflammatory phenotype in cirrhosis. Notably, T-cell receptor (TCR) activation in cirrhotic patients is associated with an increased number of HLA-DR+ T cells in peripheral blood. Although these cells coexpress programmed death receptor-1 (PD-1), their function is impaired. Further studies revealed that the enrichment of HLA-DR+CD8+ T cells in cirrhosis patients is associated with increased expression of immune checkpoint molecules (PD-1, CTLA-4, and TIM-3) on their surface, providing further evidence of T-cell dysfunction in these patients, which increases the risk of concurrent infections (43).
4. Summary and Prospects
As one of the largest visceral organs in the human body, the liver plays a crucial role in maintaining immune balance and eliminating pathogenic microorganisms. Liver diseases compromise hepatic immune function, heightening susceptibility to microbial pathogens and elevating the risk of secondary infections. Therefore, there is an urgent need to explore therapeutic strategies for secondary infections in patients with liver diseases to improve the clinical management of patients. Secondary infections in patients with liver cirrhosis remain a major clinical challenge with profound implications for patient outcomes. This review highlights that immune disorders caused by liver cirrhosis, such as elevated IL-2 levels, Kupffer cell dysfunction, immune dysfunction, and complement system defects, are crucial factors that increase the risk of secondary infections in patients. In the future, prevention and treatment strategies for secondary infections in patients with liver cirrhosis should focus on the following aspects. First, it is essential to further improve the assessment system of the immune status of patients with liver cirrhosis to achieve accurate prediction of infection risk. Second, we should explore therapeutic interventions targeting immune dysregulation by developing novel immunomodulatory drugs to restore immune balance in patients. Furthermore, we should strengthen the management of infection-related complications in liver cirrhosis patients, improve the level of early diagnosis and treatment, and reduce infection-related mortality rates. Secondary infections in cirrhosis arise from multifactorial immune dysregulation. Elucidating these mechanisms is critical to improving survival. Ongoing research, we aim to find more effective prevention and treatment strategies to improve the prognosis and quality of life of patients with liver cirrhosis.
