



1. Introduction
Metanephric adenoma (MA) is a rare benign kidney tumor with an excellent prognosis (1). The most common part of the kidney involved in various benign or malignant tumors is the tubular epithelium (2). MA originates from epithelial cells, and because of its commonly unspecific clinical and radiological characteristics, it can be difficult to distinguish it from malignant neoplasms preoperatively (3, 4). The definitive diagnosis of MA depends on the postoperative pathological findings (5). MA is derived from the residual renal organization during embryonic development (4). It is also considered to arise from different forms of maturation-arrested embryonic rests, persistent blastoma, or maturing nephroblastoma (6).
According to previous studies, about 0.2% of renal tumors are MA (7). The mean age of patients with MA is about 41 years, ranging from 5 months to 83 years, as reported in previous studies. The female-to-male incidence ratio is about 2: 1. The mean size of MA is about 5.5 cm with a range of 0.3 to 15 cm (8). MA is commonly diagnosed incidentally with no symptoms. The presenting symptoms of MA include hematuria, pyrexia, flank pain, abdominal mass, and polycythemia (9). In this study, we aimed at describing the case of a 29-year-old woman with a diagnosis of MA after nephrectomy.
2. Case Presentation
The patient was a 29-year-old woman with a history of diabetes and hypothyroidism, who was referred to the hospital emergency department due to right lower quadrant (RLQ) pain. She did not have any urinary symptoms, such as hematuria. Possible gynecological factors were ruled out by an obstetrician/gynecologist. Due to the suspicion of appendicitis, she was monitored by a general surgeon. She underwent routine laboratory and ultrasound examinations by the general surgery team. The renal ultrasound revealed a cyst with an internal echogenic content, high internal echogenic foci, and a thick wall, suggesting malignancy. Also, the ultrasound study was not in favor of appendicitis. The hospital urologist visited the patient and requested a computed tomography (CT) scan of the abdomen and pelvis, with and without intravenous (IV) contrast injection. The CT scan with contrast indicated a lesion with dimensions of 40 × 37 mm. A craniocaudal lesion (36 mm) was also seen in the lower pole of the right kidney (Figure 1).

The abdominopelvic CT scan with IV contrast
In images without contrast injection, the mass was measured at 40 Hounsfield Units (HU), and in images with contrast injection in the portal phase, it was measured at 70 HU. Due to the enhancement changes of more than 20 HU, enhancement was proposed for the lesion. According to the ultrasound, the lesion was homogeneous and contained echogenic areas. The wall thickness in the lateral section was about 3 mm (minimally thick). Overall, the findings suggested a hypovascular tumor, while no evidence of lymphadenopathy was seen. The delayed phase of IV contrast-enhanced CT scan was also performed. After 2 days of observation, appendicitis was ruled out. Regarding the laboratory tests, routine blood tests were normal, with a hemoglobin level of 12.8 g/L, a creatinine level of 1.1 mg/dL, an erythrocyte sedimentation rate (ESR) of 13 mm/h, and a glomerular filtration rate (GFR) of 83 mL/min.
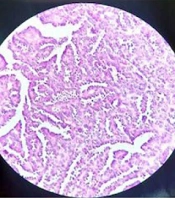
A spiral chest CT scan was carried out for the patient, which indicated normal results. The patient was considered eligible for partial nephrectomy by the urologist. However, due to extensive blood supply to the lower pole of the right kidney during surgery, she underwent a radical right nephrectomy. The nephrectomy specimen revealed a round, well-defined, tan, solid, encapsulated mass, measuring 4.5 × 4 × 3 cm in the lower pole. It did not communicate with the collective system, and no enlarged lymph nodes were detected. The microscopic sections showed a basophilic neoplasm, composed of tightly packed ducts with a scant papillary architecture. The tumor cells had a scant cytoplasm and small nuclei. Mitosis was low, and scant psammomatous calcification was seen (Figure 2). Immunohistochemically, the specimen was positive for Wilm’s Tumor 1 (WT1), vimentin, and Paired-box gene 8 (PAX8), which strongly suggested MA (Figure 3). The patient recovered completely after the surgery.

The H&E staining indicates a blue cellular tumor, composed of tightly packed, long branching, angulated ducts in a papillary architecture. Psammomatous calcification is also seen.

The positive results of IHC for WT1 and vimentin
Written and signed informed consent was taken from the patient for publishing the manuscript. This study was approved by the Ethics Committee of Shahid Beheshti University of Medical Sciences. The patient's private information remained confidential with the researchers.
3. Discussion
MA was first described by Brisigotti et al. in 1992. It has been recently classified in the group of metanephric tumors, which also include metanephric adenofibroma and stromal metanephric tumors (10). Any type of kidney tumor can be asymptomatic (11). MA is commonly asymptomatic and is found incidentally (5). Inflammatory and infectious processes, such as pyelonephritis, can delay the diagnosis of kidney tumors (12). The patient did not have any signs or symptoms of pyelonephritis. Considering the benign nature of the mass, the best surgical option for the patient was partial nephrectomy. However, since it was not possible to differentiate it from malignant masses before surgery, based on pathological examinations, radical nephrectomy is also available for the patients (13-15).
Although pathology can usually lead to a definitive diagnosis of MA, there is still an overlap between MA and papillary renal cell carcinoma (PRCC) (16). Most MA cases are well-defined, ovoid, cystic-solid, or solid renal masses with calcifications of various sizes, as well as necrotic and hemorrhagic areas (17). The ultrasound imaging results can be different for MA (hyperechoic, isoechoic, or hypoechoic) (17). In our case, the ultrasound imaging showed a hyperechoic mass. On CT scan, MA is usually well-defined with no distinct attenuation patterns; it is mostly spontaneous and slightly hyperdense compared to the normal renal parenchyma. MA may present as an isodense or hyperdense mass in CT scans with central necrosis (17). In our case, the CT scan showed a hypodense mass.
MA has some differential diagnoses, such as Wilms tumor and atypical angiomyolipoma. MA and PRCC both have homogenous densities and mild enhancements; therefore, it is difficult to distinguish between these two conditions (17). The tumor size can vary from 3 mm to 15 cm (5). The tumor is usually solid, although it can be cystic, as well (18). The tumor surface, cut by the pathologist, is usually tan to grey or yellow with hemorrhagic foci in about 20% of mass calcifications. These tumors usually lack a fibrous capsule (5). In our case, the specimen revealed a round, well-defined, tan, solid, encapsulated mass.
Microscopically, MA is composed of tightly packed, uniform, small epithelial cells with small regular nuclei, basophilic cytoplasm, a high nuclei-to-cytoplasm ratio, and no mitotic figures. The cells may be arranged in tubular or papillary architectural patterns, and glomeruloid bodies may be found. The stroma ranges from an inconspicuous to a paucicellular edematous appearance. Psammomatous calcifications are common (19). In our case, the microscopic sections showed a blue cellular tumor, composed of tightly packed, long-branching, angulated ducts with papillary architecture. Tumor cells have scant cytoplasm and small nuclei without nucleoli. Stroma is scanty and edematous in some areas; psammomatous calcification is seen, as well.
Immunohistochemical staining is also useful for diagnosing MA. It is especially useful when it is difficult to differentiate MA from PRCC. PRCC is usually positive for alpha-methylacyl-CoA racemase (AMACR) and cytokeratin 7 (CK7), whereas MA is usually positive for WT1, vimentin, and complement receptor 57 (CD57). The combination of CK7–, AMACR–, WT1+, and CD57+ is characteristic of MA (20). In our case, IHC was carried out for vimentin and WT1, which indicated positive results; however, we did not have access to AMACR or CK7.
MA has some differential diagnoses, such as PRCC and nephroblastoma. We can differentiate these tumors according to the results of IHC (Table 1) (20). CT scan and other imaging findings do not make a clear distinction between MA and its differential diagnoses. A biopsy can be used to differentiate between these tumors if there is any suspicion (17). MA is a rare kidney tumor that cannot be differentiated from PRCC solely based on clinical and radiological features, and its diagnosis is usually based on pathological findings. Therefore, further studies are needed to differentiate MA from PRCC and nephroblastoma before taking aggressive measures.
| Renal Tumor | The Results of IHC |
|---|---|
| MA | WT1 (+), CD57 (+), BRAF (+), PAX8 (+), Cadherin 17 (+), AMACR (-), CK7 (-), CD56 (-), and EMA (-) |
| PRCC | Vimentin (+), PAX8 (+), CK7 (+), AMACR (+), WT1(-), CD57 (-), and BRAF (-) |
| Nephroblastoma | PAX8 (+), WT1 (+), Vimentin (-), CD57 (-), and BRAF (-) |
Abbreviations: IHC, immunohistochemistry; MA, metanephric adenoma; PRCC, papillary renal cell carcinoma; WT1, Wilm’s tumor 1; BRAF, v-raf murine sarcoma viral oncogene homolog B1; CD57, complement receptor 57; PAX8, paired-box gene 8 (PAX8); AMACR, alpha-methylacyl-CoA racemase; CK7, cytokeratin 7; EMA, epithelial membrane antigen.
