



1. Introduction
The gastrointestinal neuroectodermal tumor (GNET) is a rare mesenchymal tumor mainly arising in the gastrointestinal tract that has been previously known as a clear cell sarcoma-like tumor of the gastrointestinal tract (CCSLT-GT) (1). The term GNET was first used in 2012 by Stockman et al. while describing 16-case series with features differing from CCS. Thus, despite sharing some features with soft tissue clear cell sarcomas, GNET was defined as a new tumor entity with specific clinical, histologic, immunohistochemistry, and molecular characteristics (2). Nevertheless, challenges remain for diagnosis, staging, treatment, and management, mainly due to the rarity of this disease and the limited to small case reports and case series having been described to date (2-4). Herein, we report a case of GNET that is a 22-year-old male, who had previously received chemotherapy for metastatic primitive neuroectodermal tumor (PNET) and subsequently developed an abdominal mass.
2. Case Presentation
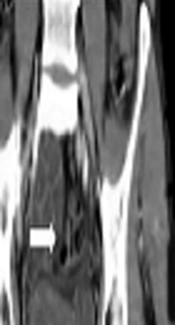
A 22-year-old male with an abdominal mass in the lower right quadrant was referred to the Shohadaye Tajrish Hospital in 2020. He had been previously diagnosed with metastatic PNET to the lung and liver and received 30 cycles of adjuvant chemotherapy. The patient did not respond to the chemotherapy. The physical examination demonstrated a palpable mass (5 × 5 cm) in the lower right quadrant of the abdomen. The CT scan of the abdomen detected an oval-shaped cystic mass with a calcified rim (55 × 40 mm) in the ileocecal region (Figure 1). The patient, then, underwent resection and appendectomy.

Figure 1.
A cystic lesion with peripheral calcification in the right lower quadrant
Cut sections of the tumor showed a solid-cystic mass. The cystic part measured 4 × 3 × 3 cm and 2.5 × 2.5 × 1 cm containing bloody fluid. The heterogeneous solid part measured 3.2 × 3 × 1 cm and revealed a yellow-white cut surface with areas of necrosis.
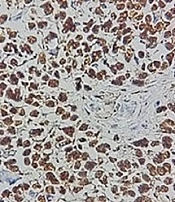
The capsuled mass was an intact cyst with some foci of ossification and calcification in the wall. Pathological examination of the ileocecal lesion revealed a solid uniform neoplastic population consisting of diffuse sheets of epithelioid tumor cells with round nuclei, surrounded by variable amounts of clear to eosinophilic cytoplasm (Figure 2A). The nuclei showed granular chromatin and occasional prominent nucleoli (Figure 2B). Tumor necrosis and increased mitotic activity (approximately 8 mitoses per 10 high power fields [HPFs]) were noted. Osteoclast-like giant cells were also identified, unevenly scattered between tumor cells. A range of immunohistochemical (IHC) markers were employed for the final diagnosis (Table 1). The IHC staining on tumor cell-block sections showed diffuse positivity for SRY-Box 10 (SOX-10), CD99, and CD56, and patchy positivity for pan-cytokeratin (pan-CK) (Figure 3). Immunoreactions against desmin, human melanoma black 45 (HMB-45), melanoma antigen (Melan-A), and chromogranin were all negative (Figure 4). The few tumor cells showed reactivity for synaptophysin (Figure 5). The molecular profile of our GNET patient was not available. Unfortunately, the patient died 1 year after diagnosis.
Table 1.Antibodies Used in the Study
| Antibodies | IHC Results |
|---|---|
| SOX10 | Diffuse positive |
| CD99 | Diffuse positive |
| CD56 | Diffuse positive |
| pan-CK | Patchy positive |
| Desmin | Negative |
| HMB-45 | Negative |
| Melan-A | Negative |
| Chromogranin | Negative |
| Synaptophysin | Low positive |
. Solid uniform neoplastic population consisting of diffuse sheets of epithelioid tumor cells with round nuclei surrounded by variable amounts of clear to eosinophilic cytoplasm (X100) (B).")
Figure 2.
Morphology of neoplastic cells and scattered osteoclast-like giant cells (A). Solid uniform neoplastic population consisting of diffuse sheets of epithelioid tumor cells with round nuclei surrounded by variable amounts of clear to eosinophilic cytoplasm (X100) (B).
 (A), CD99 (X400) (B), CD56 (X400) (C) and patchy positivity for pan-CK (X100) (D) in tumor cells The nuclei show vesicular chromatin and occasional prominent nucleoli (X400).")
Figure 3.
Diffuse positivity for SOX10 (X400) (A), CD99 (X400) (B), CD56 (X400) (C) and patchy positivity for pan-CK (X100) (D) in tumor cells The nuclei show vesicular chromatin and occasional prominent nucleoli (X400).
 (A), HMB-45 (X400) (B), Melan-A (X400) (C), and chromogranin (X400) (D) were all negative.")
Figure 4.
Immunoreactions against Desmin (X100) (A), HMB-45 (X400) (B), Melan-A (X400) (C), and chromogranin (X400) (D) were all negative.

Figure 5.
Scattered positivity of synaptophysin in tumor cells
3. Discussion
In this study, we reported a 22-year-old male patient with a malignant GNET. Gastrointestinal neuroectodermal tumor is a rare primary mesenchymal tumor of the gastrointestinal tract, with <120 cases have been reported to date, most of which were case reports or small series studies. The largest series, to date, belongs to Kandler's study, describing 20 GNET cases (3). Gastrointestinal neuroectodermal tumor is an aggressive malignancy with rapid progression and poor prognosis that occurs predominantly in the small intestine, followed by the stomach and colon (5-7). It tends to arise mainly in young to middle-aged adults with no apparent gender preference (2, 7, 8). The GNET patients commonly demonstrate gastrointestinal-related symptoms and/or abdominal masses that are detectable clinically or on imaging. However, non-specific symptoms, including high-grade fever, weight loss, anorexia, and anemia have also been reported (2, 6, 7). The GNET cases are usually prone to local recurrence and metastasis even after surgical excision (4), and metastasis at diagnosis is not unusual in GNET patients (7). Due to its rarity, variability of clinical presentation, and resemblances to a variety of other malignancies such as GIST, monophasic synovial sarcoma (SS), primary or metastatic malignant melanoma, and CCSTA (clear cell sarcoma of tendons and aponeuroses), GNET is prone to be misdiagnosed and mistreated (3). However, a thorough IHC panel with correct markers helps pathologists make more accurate and detailed diagnoses in most cases. For example, Suarez-Vilela et al. showed that GNET and synovial sarcoma, while showing overlapped immunophenotypes, can be distinguished using antibodies against CD99 and SOX10 (9). The IHC hallmark of GNET tumors is the presence of SOX-10 and S100 proteins and the absence of melanocytic-specific markers, such as HMB-45 and Melan-A (1-3, 10). The GNET patient presented here showed typical histomorphologic features of GNET, such as round to oval cells comprising nuclei with vesicular chromatin and prominent nucleoli surrounded by variable amounts of clear to eosinophilic cytoplasm, as well as variably scattered osteoclast-like multinucleated giant cells (3, 11). The mitotic rate of GNET cases varies between 1 and 30 (mostly around 10 – 12) per 10 HPFs (3, 7, 12). This observation is consistent with the case presented in this study. Consistent with the previous study (7), tumor cells showed positivity for SOX10, CD56, CD99, and synaptophysin and were negative for HMB-45, Melan-A, chromogranin A, and desmin.
Most of the GNET cases reported so far have no history of malignancy of any different kind (11). However, a few cases developed GNET after previously known malignancy (13, 14). The first case was a 15-year-old male, who developed CCSLGT virtually 15 years posttreatment with very low-dose RT (4.5 Gy) for stage 4S neuroblastoma (13). The second case was a 33-year-old male, who also developed CCSLGT 20 years after treatment with surgery, radiotherapy, and cisplatin and doxorubicin chemotherapy for childhood hepatoblastoma (14). The third case was a 33-year-old female, who developed GNET nearly 14 years after treatment with surgery and chemotherapy for gastric adenocarcinoma (11). A 22-year-old male patient with GNET presented in this study had been previously diagnosed with metastatic PNET to the lung and liver and received 30 cycles of adjuvant chemotherapy. Therefore, this would suggest that chemotherapy might be a critical risk factor for GNET development.
The Ewing sarcoma breakpoint region1 (EWSR1, previously known as EWS) gene rearrangements are the most common genomic alteration reported in GNET patients (2, 7). The EWSR1, located on human chromosome 22 (22q12), encodes a 656-amino acid protein (15), which plays a critical role in meiotic cell division, mitotic spindle formation, and stabilization of microtubules (16), as well as DNA repair mechanisms and cellular aging (17, 18). Gastrointestinal neuroectodermal tumor is associated with chromosomal translocations involving EWSR1 and one of the following partners: cAMP-dependent activating transcription factor 1 (ATF1) on chromosome 12, cAMP-responsive element-binding protein 1 (CREB1) located on chromosome 2, or less commonly, Friend leukemia virus-induced erythroleukemia-1 (Fli-1) on chromosome 11 (3, 10, 19). Therefore, EWSR1 gene rearrangements can support the diagnosis of GNET. However, we should bear in mind that chimeric EWS proteins generated by chromosomal translocations are not unique to GNET but are also found in several other soft tissue tumors such as Ewing sarcoma, myxoid liposarcoma (MLS), extraskeletal myxoid chondrosarcoma (EMC), angiomatoid fibrous histiocytoma (AFH), CCSST, CCSLT-GT, primary pulmonary myxoid sarcoma (PPMS), etc. (17). Therefore, EWSR1 rearrangement, albeit occurring often, is not a decisive diagnostic criterion for GNET. In conclusion, 3 features of gastrointestinal tract tumors that can confirm the diagnosis of GNET are as follows: (1) Containing epithelioid cells set in various patterns and/or sheets of spindle cell tumor cells, (2) strong expression of S100 and SOX10 proteins while lacking melanocytic markers expression on immunostaining, (3) and EWSR1 chromosomal rearrangement with ATF1, CREB1, or Fli-1.