



1. Background
Today, the use of bioinformatics knowledge along with experimental studies is a necessity in biotechnology research. Protein engineering, especially antibody engineering via bioinformatics methods, is one of the most important purposes for researchers (1). By using the fast and low-cost computational technique of docking, we can investigate the interaction between antibody-antigen and by identifying the effective epitopes, we can design the antigen-antibody interaction with desired characteristics (2, 3).
Cluster of differentiation 24 (CD24), also known as a signal converter and heat-stable antigen, is a cell adhesion molecule in humans. This molecule is expressed on the surface of B lymphocytes and differentiating neuroblasts and causes the production of sialoglycoprotein (4). Cluster of differentiation 24 is overexpressed in many tumors like liver, breast pancreatic, prostate, ovarian, colorectal, and renal cancer. It plays a significant role in the signaling pathways in cancer cells. Also, this marker has been used extensively for isolating cancer stem cells from solid tumors (5-7). Since CD24 is involved in the invasion, overgrowth, and metastasis of tumor cells, it could be an important marker in tumor prognosis and diagnosis (8). Because of variable glycosylation on CD24, it operates as a versatile ligand in different cancer cells with various physiological functions that complicate its mechanisms to understand (9). So, it seems CD24 can be a proper candidate in the therapeutic approaches using antibodies (10).
Many studies have investigated that the monoclonal antibodies against CD24 enhance the chemosensitivity of cancer cells. Therefore, with more studies and antibody engineering, it is possible to obtain the desired antibody against the target antigen (11). Monoclonal antibodies (mAbs) were produced by hybridoma technology in 1975. Monoclonal antibodies have a monovalent affinity and bind only to the specific epitope of the target antigen and this feature causes the antibody specificity against the desired antigen. Currently, there are many monoclonal antibodies against pathogenic antigens on the pharmaceutical market (12, 13). Single chain fragment variable (scFv) is an engineered form of a full antibody that consists of variable regions of the heavy chain (VH) and light chain (VL) and has no constant region. The chains are joined to each other by a flexible linker and comprise a single chain structure (14, 15). In recent years, antibody fragments, including single-chain antibodies, due to having multiple advantages, have been noticed by researchers for the treatment and targeted diagnosis of diseases. Also, due to their small size, rapid penetration into target cells, economical production, comfortable genetic manipulation, and high affinity to antigens could be worthy candidates in the diagnosis and treatment of diseases. Also, studies have shown that scFv is a fragmented antibody with the same antigen-binding capacity as the complete antibody (16-20).
2. Objectives
In our previous study, the scFv against CD24 was expressed and purified in vitro by the phage display technique (21), the desired scFv showed affinity to the CD24 expressing A549 cells, which was confirmed by ELISA. In this study, using bioinformatics tools, the antigenic epitopes and the structure of the scFv antibody selected by phage display were investigated. Also, after modeling, molecular docking of scFv and the CD24 antigen was done, and the important residues of antigen and heavy and light chains of scFv were identified. Therefore, this study could help us design a new and more effective antibody for therapeutic purposes.
3. Methods
3.1. Analysis of scFv and CD24 Antigen Sequence
The IMGT/V-Quest database was used to identify the scFv amino acid sequence and the complementarity-determining regions (CDRs) of the VH and VL domains (22). The CD24 antigen sequence was evaluated with the IEDB available resource (http://www.iedb.org) and the ElliPro server for detection of the potential B-cell linear and conformational epitopes (23).
3.2. Modeling of scFv and CD24 Antigen
The 3-dimensional structure of the scFv antibody was built by Sabpred Antibody Builder. In this server, heavy and light chains were submitted and, then, the initial model was made. We used Chimera software for the visualization of the 3D structure of the antibody. The 3D structure of CD24 antigen was generated, using I-TASSER online server (24). The validity of the model was evaluated by Ramachandran plots received from the (http://www.ebi.ac.uk) website.
3.3. Molecular Docking of scFv and Antigen
For determining the efficient residues involved in scFv-CD24 interaction, the Cluspro server was used (25). In other words, a 3D model of scFv was employed for docking with CD24. The Cluspro 2.0 (https://cluspro.bu.edu/publications.php) web server is used for protein-protein docking. The antibody and antigen were recognized by defining the antibody and CD24 antigen as receptor and ligand, respectively. During the analysis, all desired parameters were accepted. The masking of non-CDRs choice was selected in the antibody chains for obtaining better docking results. Thereafter, we visualized the docking result with Chimera and identified the significant amino acids involved in interactions in heavy and light chains of scFv by using the PDBsum program.
3.4. Docking Complexes Molecular Dynamics Simulation
Besides, the docked model created by Cluspro was used for molecular dynamics (MD) simulation. To investigate the reaction between scFv and CD24 in the complexes, all-atom MD simulations were performed, using the GROMACS 4.5.3 with the GROMOS96 43a1 force field. The SPC water model was used to delineate the water molecules. The initial structures were remarked scFvs in complex with CD24. Berendsen temperature coupling method was used for regulating the inside box temperature. To preserve the pressure of the system at a fixed 1 bar pressure, the ParrinelloeRahman pressure coupling method with two psbarostat time constants was used. The computing electrostatic interactions were evaluated by the Particle Mesh Ewald technique. The scFv/CD24 complex was solvated in a dodecahedron salvation box 1.2 nm from the box's edges. For neutralizing the systems Na and Cl, ions were added and extra ions were added in concentrations according to the physiological condition. After minimizing the energy by using the steepest descent algorithm, the system was stabilized at 500 ps of simulation in standard NVT and a 2000 ps simulation in the isothermal-isobaric (NPT) ensembles. The final balanced systems were subjected to 50 ns MD simulation in an isothermal-isobaric ensemble, using the leap-frog algorithm with an integration time step of 1 fs. The electrostatic and Van der Waals bonds were truncated at 10 Å. LINCS Procedure was used for checking the bond lengths in the interactions with the hydrogen atoms. In addition, root mean square deviation (RMSD), the radius of gyration (Rg), and the number of hydrogen bonds were checked to predict dynamic behavior and structural varies of the refined antibody chain. By using Chimera software, all visualization of the 3D structures was observed (26).
4. Results
4.1. Sequence Analysis of scFv and Antigen
According to the ELISA results in the experimental study, the affinity of the produced scFv antibody was 1.227 μM. By using the IMGT/V-Quest database, the VH and VL sequences of the scFv were aligned and the complementary determining regions (CDRs) and frameworks (FR) of the VH and VL domains were identified (Figure 1). The CD24 antigen sequence was submitted in IEDB resource and linear B-cell epitopes were obtained. Also, due to the protein function depending on its 3D structure, conformational epitopes were derived from PDB of antigen using the ElliPro server (Tables 1 and 2).
 and hypervariable regions (CDR) domains. Red, orange, and purple colors display CDR1, CDR 2, and CDR 3 of VH antibody respectively and black colors show FR regions. Also, blue, green, and dark green colors show CDR1, CDR2, and CDR3 in the VL antibody domain, and framework regions have also been demonstrated by black colors.")
Single chain variable fragment frameworks (FR) and hypervariable regions (CDR) domains. Red, orange, and purple colors display CDR1, CDR 2, and CDR 3 of VH antibody respectively and black colors show FR regions. Also, blue, green, and dark green colors show CDR1, CDR2, and CDR3 in the VL antibody domain, and framework regions have also been demonstrated by black colors.
| No. | Chain | Start | End | Peptide | Number of Residues | Score |
|---|---|---|---|---|---|---|
| 1 | A | 3 | 14 | GRFCPESPPGFV | 12 | 0.718 |
| 2 | A | 82 | 88 | TSSNSSQ | 7 | 0.685 |
| 3 | A | 95 | 104 | LAPNPTNATT | 10 | 0.652 |
| 4 | A | 106 | 129 | AAGGALQSTASLFVVSLSLLHLYS | 24 | 0.638 |
| 5 | A | 17 | 34 | AATSAVSLDPPSGEPRPG | 18 | 0.596 |
| 6 | A | 39 | 43 | GPRSA | 5 | 0.564 |
Acquisition of Linear Epitopes of CD24 Antigen
| No. | Residues | Score |
|---|---|---|
| 1 | A:V2, A:G3, A:R4, A:F5, A:C6, A:P7, A:E8, A:S9, A:P10, A:P11, A:G12, A:F13, A:V14, A:R15, A:V16, A:A17, A:A18, A:T19, A:S20, A:A21, A:V22, A:S23, A:L24, A:D25, A:P26, A:P27, A:S28, A:G29, A:E30, A:P31, A:R32, A:P33, A:G34, A:P38, A:G39, A:P40, A:R41, A:S42, A:A43, A:A44, A:R46, A:G49, A:C50, A:T51, A:A52, A:P53, A:A54, A:W62, A:E63, A:T64, A:K72, A:I73, A:S75, A:S76, A:T78, A:T82, A:S83, A:S84, A:N85, A:S86, A:S87, A:Q88, A:S89, A:G94, A:L95, A:A96, A:P97, A:N98, A:P99, A:T100, A:N101, A:A102, A:T103, A:T104, A:K105, A:A106, A:A107, A:G108, A:G109, A:A110, A:L111, A:Q112, A:S113, A:T114, A:A115, A:S116, A:L117, A:F118, A:V119, A:V120, A:S121, A:L122, A:S123, A:L124, A:L125, A:H126, A:L127, A:Y128, A:S129 | 0.58 |
Acquisition of Conformational Epitopes of CD24 Antigen
4.2. Modeling of the scFv and CD24 Antigen
Using the I-TASSER online server, a 3D model of CD24 antigen was built (Figure 2A). The 3D model of the scFv antibody was created by the SAbpred server. We observed the 3D model of the antibody by the Chimera program (Figure 2B).
 antibody. A, 3D model of CD24 antigen by I-TASSER server; B, visualization of the 3D model of antibody with chimera, CDR1, CDR2, and CDR3 of the VH antibody domain is red, orange, and purple respectively and CDR1, CDR2, and CDR3 of the VL antibody domain is blue, green and dark green respectively; C, Ramachandran plots of the antigen CD24 model and; D, Ramachandran plots of the scFv models.")
Bioinformatics analysis of the CD24 antigen and single chain variable fragment (scFv) antibody. A, 3D model of CD24 antigen by I-TASSER server; B, visualization of the 3D model of antibody with chimera, CDR1, CDR2, and CDR3 of the VH antibody domain is red, orange, and purple respectively and CDR1, CDR2, and CDR3 of the VL antibody domain is blue, green and dark green respectively; C, Ramachandran plots of the antigen CD24 model and; D, Ramachandran plots of the scFv models.
The antibody 3D structure and CD24 were assessed and authenticated with the Ramachandran diagrams resulting from (http://www.ebi.ac.uk) website Figure 2C and D . The antibody Ramachandran plot showed that 173 residues (91.1%) have been situated in the favored area, 15 residues (7.9%) in the additional allowed area, 1 residue (0.5%) in the generously allowed area, and 0.5% of the residues were in disallowed areas. Also, in the antigen Ramachandran plot, 54 residues (54%) in the favored regions [A,B,L], 20 amino acids (20%) in additional allowed regions [a,b,l,p], 17 amino acids (17%) in generously allowed regions [~a,~b,~l,~p], and 9 residues (9%) in disallowed regions have been located.
4.3. Molecular Docking of CD24 Antigen and scFv Antibody
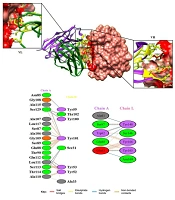
Molecular docking between CD24 and the scFv was calculated by ClusPro 2.0 to determine the interactions of CD24 and the scFv model. The greatest model with the lowest energy was chosen among 10 models. The models were built and visualized by the Chimera program (Figure 3A). After docking, the important amino acids in the VH and VL chains of scFv were determined with the PDBsum program (Figure 3B). The obtained results were shown 9 and 6 hydrogen bonds between H and L chains with CD24 antigen, respectively.
 antibody and cluster of differentiation 24 (CD24) antigen from the Chimera analysis. The docking complex between the antigen (pink surface) with VL (green ribbon) and VH (purple ribbon) is shown. Yellow and red residues belong to the scFv and epitopes of the antigen, respectively; B, Amino acids involved in the antigen–heavy chain (VH) and antigen–light chain (VL) complexes. Salt bridges, disulphide, hydrogen bonds, and non-bonded contacts of antigen-antibody complexes are shown.")
A, 3D structure of the docking complex between single chain variable fragment (scFv) antibody and cluster of differentiation 24 (CD24) antigen from the Chimera analysis. The docking complex between the antigen (pink surface) with VL (green ribbon) and VH (purple ribbon) is shown. Yellow and red residues belong to the scFv and epitopes of the antigen, respectively; B, Amino acids involved in the antigen–heavy chain (VH) and antigen–light chain (VL) complexes. Salt bridges, disulphide, hydrogen bonds, and non-bonded contacts of antigen-antibody complexes are shown.
4.4. Molecular Dynamics Simulation of scFv and CD24 Antigen Complex
After docking, we applied the GROMACS 4.5.3 package for the simulation of the intended docked scFv and CD24 antigen complex. The RMSD, RG, and number of hydrogen bonds were determined to assess the stability status of complexes during simulations. Figure 4A depicts the RMSD values of the complex during the simulation period. After 30 ns of simulation, the RMSD of the scFv-CD24 antigen reached a plateau, indicating system equilibration. RG analysis was employed to consider the unfolding or folding position of the complex. The low variations in RG displayed high stability of the complex. Figure 4B shows the stability of the docked complex after 30 ns. The number of H-bonds between scFv and antigen (CD24) were analyzed and the results are illustrated in Figure 4C.
, value of scFv- CD24 antigen complex; B, Rg value of scFv- antigen CD24 complex; and C, The number of hydrogen bonds between VH and VL chains of single chain variable fragment (scFv) with CD24 antigen.")
A, Root Mean Square Deviation (RMSD), value of scFv- CD24 antigen complex; B, Rg value of scFv- antigen CD24 complex; and C, The number of hydrogen bonds between VH and VL chains of single chain variable fragment (scFv) with CD24 antigen.
5. Discussion
Computational biology is one of the most effective tools for studying protein-ligand binding mechanisms and investigation of antigen epitopes involved in antibody binding in this interaction. Available bioinformatics software and tools play a significant role in designing more effective antibodies in the targeted treatment of many cancers.
In a study in 2020, the antibody was prepared in scFv format, and amino acids effective in binding antigen and antibody were identified by molecular docking. The virtual amino acid mutation was performed to improve scFv affinity. The results showed that finding amino acids involved in antigen-antibody interaction can be used to modify scFv properties in vitro (27).
In another study, a scFv antibody against salbutamol (SAL) was prepared, using the phage display method. Then, using the docking results, scFv with high affinity to SAL was obtained, which can be used for rapid and inexpensive diagnosis of SAL (28).
In our previous experimental study, a specific scFv antibody against CD24 antigen with appropriate affinity was isolated, using phage display (21). In this in silico study, the 3D structure of CD24 antigen and antibody were predicted. By antigen-antibody complexes analysis from docking, the important amino acids involved in these interactions were recognized.
Fifteen hydrogen bonds were observed between CD24 antigen and antibody residues, of which only 3 residues (Tyr59, Tyr165, and Asn169) were located in the FW region and the other 12 residues were located in the CDRs region. Tyr148 in CDR 1 of the VL chain has 4 hydrogen bonds with the CD24 antigen. Also, 9 hydrogen bonds were observed in the VH chain and 6 bonds in the VL chain. Due to the higher number of hydrogen bonds between the heavy chain's amino acid residues with the antigen compared to the light chain, a new nanobody with higher affinity can be designed by using the scFv heavy chain. However, more experimental and in silico analyses are necessary in this field.
In this regard, there are similar studies. Using an in silico technique, Ebrahimi et al. altered the affinity of the PLGF nanobody (17). They identified the critical amino acids in the nanobody and subsequently developed numerous point mutations that improved the affinity to PLGF. High-affinity nanobody variants were generated based on their evaluations, which were based on docking and MD simulations.
According to another study, researchers predicted and confirmed the VH antibody fragment interacting with factor H binding protein from Neisseria meningitidis (29). They simulated the 3D model of the VH fragment by online server. Using the ClusPro 2.0 server, at first, the 3-dimensional structure of the related antigen was docked to the VH fragment model. GROMACS 4.5.3 was used for MD simulation. Secondly, the VH fragment sequence was cloned and expressed. Then, its binding activity to the fHbp protein was determined, using an ELISA assay and the surface plasmon resonance (SPR) method. The results confirmed the importance of the residues implicated in the fHbp-VH interaction discovered in silico.
Bandehpour et al. constructed the 3D structures of 2 scFv antibodies by Kotai antibody builder. Then, the scFv models and Mycobacterium ESAT-6 antigen were docked by using the ClusPro 2.0 web server, and MD simulations were performed. Significant amino acids involved in ESAT-6antigen-antibody complexes were defined, which belonged to the CDRs, including VL3 Asn164, VL7 Ser164, and VH7 Asn55. So, based on these results, designing new antibodies with affinity enhancement can proposed for tuberculosis diagnosis (30).
5.1. Conclusions
In this study, a computational method was applied for the determination of critical amino acids of CD24 epitopes in the binding with the scFv by docking steps. Then, the best docking structure of antibody-CD24 antigen was purposed for MD simulation steps that energy screening of the complex was measured. In silico achievement proposes other substitutions for targeted therapy by determining and alternating involved crucial amino acids in interaction and designing new scFv with more effective function to antigen, which would be helpful for immunotherapy.
