




1. Introduction
Paragangliomas (PGLs) are vascular tumors associated with blood vessels and neural structures (1). About 80% of these tumors occur in the adrenal medulla often termed pheochromocytoma (PCC) (2) and the rest occur at extra-adrenal locations. Depending upon location, functional status of the tumor and some other factors, the clinical manifestation of PGLs may differ with some patients even being asymptomatic. Given vague clinical presentation, reaching the diagnosis of a PGL can sometimes be challenging and, thus, its early diagnosis requires a high index of suspicion.
2. Case Presentation
A 17-year-old female patient was referred to our emergency department with a sudden blurred vision along with palpitation for 1 week. She had a 2-month history of nausea and a vague, diffuse, and non-radiating abdominal pain. The patient had also experienced occasional restlessness, flushing, and headache for about 1 year. She did not notice any alteration in defecation habits. The medical history, as well as family history, was clear and the patient denied any drug or substance abuse. On the physical examination, she was found to be thin, tachycardic at 110 bpm, and hypertensive with a blood pressure of 190/110 mmHg. Abdominal examination was insignificant other than a mass detected on the left side of the umbilicus with mild tenderness on deep palpation. The electrocardiogram showed tachycardia with normal sinus rhythm and no abnormal changes. An emergent ophthalmology consultation revealed dilated conjunctival vessels and hypertensive retinopathy. The patient underwent spiral abdominal and pelvic computed tomography (CT) scan, which showed a highly hypervascular retroperitoneal mass in the left infrarenal region with a heterogeneous enhancement pattern, tightly fixed to the aorta, surrounding the inlet of the aortic bifurcation, and the left iliac artery measuring 12 × 6 × 6 cm (Figure 1). Based on the information provided, the main differential diagnoses consisted of malignant vascular tumors, hypervascular metastases, and extra-adrenal PCCs.
 with a heterogeneous enhancement pattern surrounding the inlet of the aortic bifurcation and the left iliac artery.")
A, Coronal; and B, axial section of the CT scan showing a mass (arrows) with a heterogeneous enhancement pattern surrounding the inlet of the aortic bifurcation and the left iliac artery.
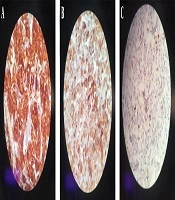
Laboratory testing showed a normal vanillylmandelic acid (VMA) level, mildly-elevated urine metanephrine (1.3, normal range: < 1.3), and urine normetanephrine (82, normal range: 15 - 80) suggestive of a PGL. So, I-131 metaiodobenzylguanidine (MIBG) scan was done, which depicted a curvilinear-shaped area in the abdomen and a large area of increased radiotracer uptake in the pelvic region, suggesting intestinal mesentery involvement by a tumor with a neuroectodermal origin. The patient was pretreated with 20 mg of phenoxybenzamine TDS and once stabilized, an exploratory laparotomy was performed through the left retroperitoneal approach. Vascular control was obtained at the infrarenal aorta and, then, at bilateral iliac arteries. The left ureter was explored and the tumor was cleared off it. Exploration of the renal artery and vein was done and revealed that the tumor was located far from them. A large highly vascular retroperitoneal mass fixed to the distal abdominal aorta was evident, which was cleared off the aorta, and the arterial branches were ligated. The tumor was completely resected without any detriment to the aorta and the need for vascular reconstruction. Exploration of the peritoneal cavity revealed no tumor involvement. The immunohistochemistry (IHC) staining of the specimen was positive for synaptophysin and chromogranin, and sustentacular cells showed positivity for S-100 protein (Figure 2). Histopathological examination confirmed the lesion to be a benign PGL measuring 11 × 9.5 × 9 cm. There was also peripheral margin involvement, as well as capsular and perineural invasion without any lymphovascular invasion. Two examined dissected lymph nodes were tumor-free. The patient had an uneventful postoperative course. At the most recent follow-up, which was approximately 3 years after the surgery, she was found to be hypertensive. Laboratory data showed elevated plasma normetanephrine (770.1 pg/mL, normal range < 196 pg/mL), plasma norepinephrine (1730 pg/mL, normal range up to 600 pg/mL), 24-hour urine normetanephrine (1232 Ug/24h, normal range < 600 Ug/24h), and 24-hour urine norepinephrine (129 Ug/24h, normal range < 90 Ug/24h) levels. MIBG single-photon emission computed tomography (SPECT) scan revealed avid nodular uptake in both lungs, more prominent on the right side (Figure 3), compatible with tumor recurrence. The patient reported no respiratory complaints. The abdominal region was clear from the tumor on synchrotron radiation -CT (SR-CT) scan (Figure 4) and MIBG scan.

IHC staining with the original magnification of 40× showing positivity for A, synaptophysin; B, chromogranin; and C, S-100.

A, Coronal; and B, transverse sections of MIBG SPECT-CT scan showing avid nodular uptake in both lungs, more prominent on the right side.
 scan.")
The abdominal region was clear from the tumor on the synchrotron radiation CT (SR-CT) scan.
3. Discussion
PGLs are rare neuroendocrine tumors with high morbidity and mortality due to their multifocality and progression (3). They carry a malignancy rate of about 10% and a 5-year survival rate of < 50% (4, 5).
The classic triad of headache, sweating, and palpitations/tachycardia is uncommon, but when present, it increases the specificity to about 90% (6). Catecholamine hypersecretion may be intermittent or continuous and is usually paroxysmal.
The diagnosis of a secretory PGL is usually made through measuring urinary and/or plasma fractionated metanephrines and catecholamines. It is worth noting that biochemical testing should be done in all PGL cases, even if clinically found to be nonfunctional. As other neuroendocrine tumors, elevated serum levels of antisera to neuron-specific enolase (NSE), chromogranin A, or vimentin can be useful to distinguish these tumors from the non-neuroendocrine ones (2).
Radiologic imaging also plays an important role in the evaluation of a PGL. Among available modalities, ultrasound, triple-phase helical CT, magnetic resonance imaging (MRI) with gadolinium-diethylenetriaminepentaacetic acid infusion (7), angiography, and radioisotope imaging using MIBG and 18F-fluoro-2-deoxyglucose positron emission tomography (FDG-PET) are commonly used. Histological examination reveals a thin capsule with polygonal or round epithelioid cells arranging in compact cell nests or trabecular patterns known as Zellballen (8). On IHC evaluation, chief cells usually show positivity for neuroendocrine markers such as synaptophysin, chromogranin, and NSE, and sustentacular cells are S-100 positive (2, 8).
Due to the catecholamine crisis leading to severe hypertension, an incisional or fine needle biopsy is contraindicated in a patient with suspected PGL unless there are negative biochemical testings or alpha-adrenergic blockers are prescribed. Furthermore, a biopsy can lead to severe hemorrhage or fibrosis with subsequent difficulty with definitive surgery (1, 9, 10).
Locoregional PGLs can be managed by surgical resection or radiation therapy. Generally, the preferred approach for localized PGLs below the neck and all catecholamine-secreting PGLs at any site is resection. Laparoscopic resection is often preferred, but with large (> 6 cm) tumors or tumors with a high risk of malignancy, exploratory laparotomy is suggested (11).
3.1. Conclusions
The diversity of manifestations and rarity of PGL can delay the diagnosis, which may lead to potential complications. This makes it compelling to include PGLs in differential diagnoses in a clinical setting compatible with catecholamine hypersecretion.
