


1. Introduction
Online Mendelian Inheritance in Man (OMIM #308300), also referred to as Bloch-Sulzberger syndrome, is a rarely diagnosed x-linked Incontinentia pigmenti (IP) (dominant disease predominantly among females). The condition is lethal in males due to spontaneous abortion of the affected males, affecting approximately one in 40,000 newborns (1). It is secondary to mutations in the IKK-gamma gene, which is also known as the NF-kappa-B essential modulator that is located on Xq28 (2).
This systemic disease affects tissues with the ectodermal and mesodermal origin, including the skin, teeth, eyes, hair, and the central nervous system (CNS). Approximately 80% of the patients exhibit some other anomalies affecting the teeth, skeleton, and nails, with microcephaly, seizures, psychomotor disorders, strabismus, atrophy of the optic nerve, detachment of the retina, cataract, and some other anomalies (3, 4).
The diagnosis of IP predominantly relies on clinical features, especially integumentary manifestations (i.e., skin and its appendages). Besides, IP is characterized by erythematous eruptions with linear vesiculations, which are present at birth or appear soon after birth and are mainly localized on the skin of the back, torso, or extremities (5). Cutaneous eruptions along the lines of Blaschko are the hallmark of IP and develop in four different stages (6):
Stage 1: Inflammatory, erythematous, vesiculobullous lesions, usually linear in pattern (from birth to one or two weeks of age)
Stage 2: Papules and verrucous lesions with hyperkeratosis (two to six weeks of age)
Stage 3: Hyperpigmentation of the skin (three to six months of age)
Stage 4: Hypopigmentation and atrophy of the skin (in the second to third decades of life)
Pigmentation gently fades and is usually absent in adulthood. The affected infants are not usually systemically compromised despite eosinophilia and leukocytosis on peripheral blood count (7).
Apart from integumentary manifestations, patients diagnosed with IP might exhibit dental anomalies in both deciduous and permanent teeth (8, 9), with hypodontia as the most commonly occurring dental anomaly, detected in 43% of the cases, followed by conical, peg-shaped teeth, delayed eruption, hypomineralization, supernumerary teeth, and supplementary cusps. A vast majority of dental manifestations in patients diagnosed with IP can be managed successfully. Some of such anomalies adversely affect the patient’s Quality of Life (QoL), leaving the patient with psychological disorders as well as feeding and masticatory problems (10).
This article constitutes a report of a young girl, age 11 years, exhibiting different manifestations of IP with typical dental findings. Due to the rarity of this disease, discussing various manifestations and oral treatment helps dentists to better diagnose and treat similar cases. This case report aims to provide a viable treatment protocol combining various treatment modalities to restore the function and esthetic appearance and safeguard the patient’s dental health.
2. Case Presentation
An 11-year-old girl was admitted to the Department of Pediatric Dentistry, School of Dentistry, Isfahan University of Medical Sciences, with a chief complaint of “abnormal dentition and ugly teeth.” Written informed consent was obtained from the patient’s parents to publish this report and the relevant images. The family history was negative for any relatives with IP.
She was born to a 20-year-old mother, full-term after an uneventful pregnancy, without any complications. The child was born in a hospital (with a caesarian section) without any complicating factors. After the birth of the patient, the mother had one miscarriage involving a male fetus. The mother denied any history of skin lesions, and at the time of examination, she had no visible abnormalities on her face, neck, arms, and nails.
Eight days after birth, the patient developed pigmented vesiculobullous eruptions that first affected the scalp and then the trunk (sparsely) and limbs (predominantly on the right side). Due to this condition, the baby was admitted to the hospital for one month, and some laboratory tests were carried out. Hematologic tests yielded normal results. Routine blood tests for inflammation, infections, and autoimmune diseases yielded negative results. The C-reactive protein was normal. A biopsy from the base of a bulla showed neutrophilic infiltration and little cellular debris without cytopathies and herpes inclusion bodies. Skin lesions became purulent afterward, becoming crusted and darker. Then, vesiculobullous lesions developed again in these areas until six months of age. After six months, the affected areas were replaced by dark brown lesions, leaving linear pigmentation.
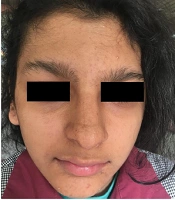
Currently, the patient has areas of hyperpigmentation on the scalp, face, trunk (sparsely), and limbs (predominantly on the right side) (Figure 1). She has developed some scarred, elevated hyperpigmented areas on the fingers of her hands (mostly the right hand), with no nail abnormalities. Her hair is of normal color and texture, but there are some areas of alopecia. Sparse eyebrows and prominent lips can also be seen (Figure 2).

Hyperpigmented lesions over the trunk and extremities

Frontal view of the patient’s face; clinical view of the hair with alopecia
The musculoskeletal system was abnormal on the left side, causing her to limp during walking. After she got older, poor eyesight (-5 eye number) and amblyopia were diagnosed. The early diagnosis of incontinentia pigmenti in this patient was possible to made by considering the vesiculobullous lesions that occurred at birth and were followed by hyperpigmented lesions that overlapped the areas previously affected by vesicobullous lesions. Gene analysis confirmed the above diagnosis by the patient being heterozygote for exons 4 - 10 deletion of the NEMO gene. Her mother also had the same mutation, but she has not had any clinical manifestations of IP. Extra- and intra-oral examinations, including panoramic radiographs, were completed (Figure 3 and 4), demonstrating hypodontia, conical teeth, and delayed tooth eruption. Soft tissue examination revealed mild gingivitis secondary to poor oral hygiene and pigmentation on the gingiva (both lower and upper arches). A narrow and atrophic ridge, especially in the posterior area of the lower arch, was seen.

Clinical view of the maxillary and mandibular arches at age 11

Panoramic radiograph showing hypodontia at age 11
Examination of the teeth showed the presence of only eight primary teeth, with carious lesions in all the primary molars and canines. There was no evidence of the presence or absence of other deciduous teeth. Only according to the patient’s mother, there were anterior deciduous teeth that were exfoliated at the age of 8.5, and the patient had no experience of tooth extraction or dental trauma causing tooth loss before referring to us. Multiple permanent tooth germs (#14, #12, #22, #31, #34, #35, #37, #41, #42, #44, #45, and #47) congenital missing were also found on the panoramic radiograph. The maxillary left and right second permanent molars exhibited delayed development. There were some interdental spaces due to some missing teeth. Based on her previous dental document, hypoplastic/hypocalcified permanent first molar teeth were restored with stainless steel crowns (SSC) in two dental sessions at the age of eight. No clinical or radiographic signs were observed related to crowns after three years of treatment, and only mild gingivitis was seen around the crowns, which was associated with mild general gingivitis due to poor oral hygiene. There were no symptoms of pulp pathology in our patient. The patient had a class I molar occlusal relationship on both sides.
In the comprehensive treatment plan, oral hygiene instructions, extraction of all unrestorable primary molars, and composite filling of all primary canines were scheduled. Therefore, to resolve the masticatory and esthetic appearance problems due to the congenital missing of several teeth, a removable space maintainer was fabricated for the patient with pontics replacing the missing primary molars in the lower jaw (Figure 5). The gingiva condition was assessed two weeks after the beginning of treatment interventions and oral hygiene instructions. Then, during the next sessions, at the same time as removing decayed teeth or repairing them, hygienic instructions were reminded, and the condition of the soft tissue of the mouth was evaluated. A dental prosthesis was delivered to the patient about three weeks after removing decayed teeth and healing of gingiva. Furthermore, after delivery of the dental prosthesis, the teeth and soft tissue were evaluated at regular intervals of one day, one week, and one month.

Lower jaw removable space maintainer containing pontics of the missing primary
Subsequently, the patient was placed on a regular check-up scheduled every six months to apply fluoride and reinforce oral hygiene instructions to prevent caries. Currently, after 22 months, the oral health and clinical outcomes of the patient are quite favorable. The parents were informed of the possible future dental treatment options, consisting of regular preventive procedures every six months to maintain the orodental health. As the skeleto-occlusal relationships might be affected by the growth spurt at puberty, implant therapy has to be delayed until after the growth spurt, combined with full-mouth orthodontic–prosthodontic procedures to relocate and align teeth during adulthood to achieve better results of full-mouth rehabilitation.
3. Discussion
Incontinentia pigmenti is a rare hereditary x-linked disease that affects the skin and teeth and results in ophthalmologic and neurologic disturbances. It might be fatal to the majority of male embryos that are affected. Telltale integumentary manifestations and anamnestic heredity data of IP help easily diagnose it in neonates. The broad range of integumentary manifestations and possible dental, neurologic, and ocular abnormalities help reach a diagnosis in older children and adults (10). According to the criteria presented by Landy and Donnai (11), cutaneous lesions (erythema, hyperpigmented streaks, and whorls, pale-appearing, hairless, atrophic lines, or patches) were categorized as essential diagnostic criteria for IP.
In contrast, dental, retinal, hair, and nail abnormalities are considered minor criteria for IP. In the absence of ophthalmic and central nervous system involvement, the physician in charge monitors and manages the cutaneous and dental manifestations if required. The prognosis of IP is generally good, depending on extra-cutaneous involvement, which might also affect patients’ QoL.
In the case presented here, IP had a familial etiology. Her mother had the same NEMO gene defect. This 11-year-old female child had typical IP skin changes in the vesiculobullous stage when she was a newborn baby. The IP diagnosis was established based on typical skin manifestations, which were confirmed later by NEMO mutation. A few years later, amblyopia and retinopathy were noted. According to the updated IP diagnostic criteria (4), she met one major and one minor criterion with NEMO mutation. Clinical expressions of IP vary from one patient to another. Her mother had the same NEMO mutation without any problem.
Virtually half of the IP cases exhibit a positive family history of the same condition. The condition has a definite female predilection (F:M = 37:1). The female patients survive due to the lyonization phenomenon (or X chromosome inactivation) during the early embryogenesis period (12). The male patients might die due to the NEMO mutation in the embryonic period, which can be the etiology of spontaneous abortion of the male embryo of our case’s mother. Less than 3% of cases are diagnosed in males, with many exhibiting Klinefelter’s syndrome (47, XXY karyotype), in which the second X chromosome plays a crucial role in their survival in utero. Different genetic mutations have been reported in some male cases, consisting of hypomorphic alleles or somatic mosaicism for common IKBKG deletion (13, 14).
Not all four stages of cutaneous manifestations of IP might be present in some cases. Stage 1 (inflammatory or vesicular) and stage 3 (hyperpigmented) are more frequent than stage 2 (papular or verrucous) and stage 4 (hypopigmented or atrophic) (8). In the present case, only stages 1 and 3 were observed. Considering the patient’s age, 11 years, she may still develop stage 4, in which even the lesions might disappear completely. As a result, while cutaneous lesions are one of the most critical factors to reach a diagnosis of the condition, they give rise to limited damage, requiring no therapeutic intervention.
Concerning frequency, orodental anomalies constitute the most important minor criteria for IP. In a systematic review by Minić et al. on the available published data during 1993–2010, orodental anomalies were found in 54% of the patients diagnosed with IP. The majority of the anomalies (95%) were dental, with the rest (5%) being oral. The most frequent dental anomalies comprised anomalies in tooth shapes and hypodontia, constituting 36% and 31%, respectively, of all the detected dental anomalies. Delayed tooth eruption was reported in 18% of the patients, with 14% of anomalies being unclassified. Of 23 different types of oral anomalies in patients diagnosed with IP, the most frequent entities were cleft palate and high-arched palate (10), which might be categorized as minor diagnostic criteria for IP. Dental and oral anomalies often influence the QoL of IP patients. Unlike other clinical findings in IP, the majority of dental and some oral anomalies could be corrected. A dentist diagnosing IP in a patient has therapeutic and advisory roles for the patient and the family (10).
The absence of teeth and significant anomalies in teeth shapes might affect normal facial development, mastication, speech acquisition, appearance, and self-esteem in the growing child (15). In childhood, parents should be provided with information about the possibility of a delayed eruption, missing teeth, and the effects on the developing dentition. It is also important to ensure that nutritional needs are being met (16). Thorough oral hygiene and healthy eating habits should be encouraged to help preserve the existing dentition for as long as possible (17).
