



1. Background
Ectodermal dysplasia (ED) is a large heterogeneous group of inherited disorders characterized by ectodermal origin appendages defects (1, 2). Faulty ectodermal development leads to defects in ectoderm‐derived structures such as hair, nails, teeth, and sweat glands. Hypohidrotic ectodermal dysplasia (HED) is the most common type of ED characterized by triad symptoms, including sparse hair (hypotrichosis), anodontia or hypodontia, and hypohidrosis oranhidrosis (3, 4). It also includes dryness of the skin, eye, and airways, maybe due to aplasia or hypoplasia of sebaceous glands and mucous membranes. The skin is thin and hypopigmented, whereas the skin around the eyes may be hyperpigmented (5-8). HED patients may seem prematurely aged due to wrinkled skin (9-11). Mental defectiveness reported in some groups could be the result of febrile seizures due to inability or insufficiency sweating, which could even threaten their lives in severe cases (12).
Mutations in ectodysplasin A (EDA) gene account for 90% of all HED cases and cause X-linked HED (13-15), while mutations in EDA-receptor (EDAR) and EDAR-associated death domain (EDARADD) genes cause rare autosomal dominant and recessive forms. It was recently shown that other genes like tumor necrosis factor receptor associated factor 6 (TRAF6) and wingless-type MMTV integration site family, member 10A (WNT10A) could also be involved in causing HED (16, 17).
The prevalence of XL-HED is estimated at 1 in 100,000 live male births, and the carrier incidence is around 17.3 in 100,000 women (18).
The interaction between EDA, EDAR, and EDARADD, which are ligand, receptor, and protein adaptor, respectively, results in normal triggering of EDA-EDAR signaling pathway. Deficiencies in the EDA-EDAR signaling pathway cause HED (19).
EDA gene, which encodes ectodysplasin, consists of eight exons and is located on long arm of chromosome X (Xq12-q13.1). The two longest isoforms of EDA are ectodysplasin-A1 and -A2, which differ by an insertion of two amino acids. EDA-A1 binds only to EDAR, while EDA-A2 isoform employs X-linked EDA receptor (XEDAR) (3). EDA-A1 is a type II transmembrane protein with 391 amino acids and a TNF superfamily member involved in primary mesenchyme-epithelial interactions and regulates the development of ectodermal derivatives with the role of embryonic morphogenesis (20). A variety of mutations in the EDA gene have been identified as missense and nonsense mutations, as well as small deletions. More than 200 mutations have been reported for EDA gene, most of which are missense. About 80% of mutations identified in EDA are small changes that include point mutations and deletions. More than half of these mutations have been seen in exons 1, 3, and 5 (1).
EDAR is a type I transmembrane receptor protein with 448 amino acids which belongs to TNF receptor superfamily and consists of an extracellular region (in N terminal), a transmembrane region, and a death domain in the intracellular region (in C terminal) (11, 19). EDAR gene is composed of 12 exons and located in chromosome 2 (2q11-q13). To date, more than 50 mutations have been identified in EDAR gene. Most of these mutations occur in exon 12, where death domain is located (http://www.hgmd.cf.ac.uk/).
2. Objectives
In this study, we investigated two distinct families with HED in southwest of Iran. Accordingly, for the first time, we identified the same mutations in EDA gene in two affected brothers in family 1 and a novel mutation in EDAR gene in an affected boy in family 2.
3. Methods
In this study, HED patients referred to Narges lab Ahvaz, Iran, in 2017 were included after clinical examination by specialists. All patients were diagnosed as HED cases based on having at least two of the three ectodermal abnormalities of teeth, hair, and sweat glands. We studied three HED patients in two distinct families in the southwest of Iran to assess gene mutations in the EDA and EDAR genes. Moreover, 120 healthy individuals were recruited as normal controls to rule out genetic polymorphism.
In this study, 5 mL of peripheral blood was collected from the probands, associated parents, and 120 matched unrelated controls (from related ethnicity) without any ectodermal disorder. DNA was extracted using the routine salting-out protocol from blood leukocytes. Primer sequences for all exons of EDA (NM_001399.4) and EDAR (NM_022336.3) and flanking intronic sites of EDA and EDAR were designed using Oligo 6.0 (Tables 1 and 2).
| Primer | Sequence | Primer | Sequence |
|---|---|---|---|
| EDAR-E1-1-F | TAGCACTCCCACGCCTAAAG | EDAR-E5-1-F | CTGCACTCACCTGTGTCCC |
| EDAR-E1-1-R | GAGGTCAGCTTCAGTCCCAG | EDAR-E5-1-R | GCAGTATCCATGACCCCTGT |
| EDAR-E1-2-F | TACCTGCTTCTGAGAGGGCT | EDAR-E6-1-F | GCTGTCCAGCTCACCTGTCT |
| EDAR-E1-2-R | GGCCACAGCTGAAGAGTTTAG | EDAR-E6-1-R | CGTGCTGAACAAATACCGTG |
| EDAR-E2-1-F | CTGCAGAGTAGGAGGCACG | EDAR-E7-1-F | CAGCGTCTTTGAAGGTGGAG |
| EDAR-E2-1-R | ATGACGTGAAGAGGCGGTAA | EDAR-E7-1-R | GCTTTCATCCGAGTACCACC |
| EDAR-E3-1-F | ATAAAGGTAGCCAGACCCCC | EDAR-E8-1-F | TCCTGAGCTCTGGGTCATTC |
| EDAR-E3-1-R | AGAGGCCAAGAAACAGTCCA | EDAR-E8-1-R | GTGAGAGCAGAAGCAGCGAG |
| EDAR-E4-1-F | GGGTCTTGCAGGAGTCACAT | EDAR-E9-1-F | CAGAGCCATGAGCCAATTCT |
| EDAR-E4-1-R | ACAATGCCACAAGCAGGAG | EDAR-E9-1-R | TGCTCAGGATCCACAGGAC |
| EDAR-E10-1-F | TGGACAAGTTCACCACAGGA | ||
| EDAR-E10-1-R | GAGCCACATCTCACAGCTCA | ||
| EDAR-E11-1-F | CCCCTTTCTGGAGATGAAAA | ||
| EDAR-E11-1-R | CTCAGTTCCCCTCACAGGAG | ||
| EDAR-E12-1-F | TTGACTGTGACTTGCAACATCT | ||
| EDAR-E12-1-R | CAGTCTTTTGGCACCACTCA |
| Primer | Sequence | Primer | Sequence |
|---|---|---|---|
| EDA_E1_1-F | GCAGAGGCGAACCCTCAC | EDA_E10_1-F | TTGGGGAAATTCTCATGGAT |
| EDA_E1_1-R | CGCAACTCTAGGTAGCAGCA | EDA_E10_1-R | TTGTTTTATAAAGCCAATGTTTGTT |
| EDA_E2_1-F | GGGCTATTTGGAGTATGTTCCT | EDA_E11_1-F | GGCTATGACTGAGTGGGGTC |
| EDA_E2_1-R | AGATCCCACCTCCCTAAAAA | EDA_E11_1-R | GCACAGTCTCAGCTCACTGC |
| EDA_E3_1-F | TTGGGACTTTGGAGGTGTTT | EDA_E12_1-F | TCTTGGGGATCCCTCCTAGT |
| EDA_E3_1-R | CCACGTGGTTAAGGCTCTTC | EDA_E12_1-R | ATACAAAAATCGCACTCTTGATT |
| EDA_E4_1-F | TGAATGACAGACTTGCTGGG | EDA_E13_1-F | AGCAGGACTCCGTCTCAAAA |
| EDA_E4_1-R | TCATCAAACTGTCAAAAGTCAAAGA | EDA_E13_1-R | GCTCTCAGGATCACCCACTC |
| EDA_E5_1-F | TGCTGCAAAGTTTAATGTCCA | EDA_E14_1-F | TTATGCCCTCTGATTGTCCT |
| EDA_E5_1-R | AAAAATGGATCACAGACCTAAATG | EDA_E14_1-R | GAAAACCGTCAGAATCTCCG |
| EDA_E6_1-F | TCATGGCCTTTCTTCCTAAGTT | EDA_E15_1-F | GATGGAAACATGGGACTGGT |
| EDA_E6_1-R | AAGTGGCACAGATGCTGGTA | EDA_E15_1-R | ATTATTTGGAGGCTGGGGAG |
| EDA_E7_1-F | TTCCCAGTCCAGTACTCTTCATC | EDA_E16_1-F | GGGGTTGTGAACTCCTTGGT |
| EDA_E7_1-R | TCTCACAAAAACAGTAAAAGGGAA | EDA_E16_1-R | CCGGATCTGCATTCTGGATA |
| EDA_E8_1-F | GCCAGCATTGGTTCTGAAGT | EDA_E17_1-F | TCTCTTCCCCAATCCCTTCT |
| EDA_E8_1-R | TGTCAACGGAAAGTTATCTGTGTT | EDA_E17_1-R | TTGTCACCCTGGAGTCACTG |
| EDA_E9_1-F | CCCCAATTCCATACCAGCTA | ||
| EDA_E9_1-R | TGTCAATGGTAAACCTGCGA |
Polymerase chain reaction (PCR) was performed for EDA and EDAR genes with 80 - 90 ng genomic DNA, 12.5 µL Master Mix (Amplicon Co.), and 1 µL of each primer. The amplification condition for each PCR reaction of EDA was 95°C for 5 min, followed by 35 cycles of 95°C for 1 min, the indicated annealing temperature for 30 s, and 72°C for 45 s, with a final extension at 72°C for 5 min. The amplified PCR fragments were separated on 1% agarose gels. Both DNA strands were sequenced using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems 3500 DNA Analyzer). Finally, chromatograms were read and analyzed by Chromas and DNA Baser v4 software.
4. Results
In this study, two families with three patients were included. Family 1 was an Arab family, which had two affected sons with clinical features suggestive of HED, including deformity of teeth, hair sparseness, and heat intolerance; this family was tested for EDA mutation. Family 2 was a consanguineous Persian family (first cousins) who had an affected boy with teeth abnormality, heat intolerance, and skin dryness symptoms; this family was tested for EDAR mutation.
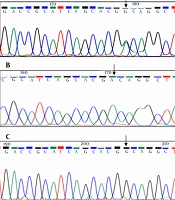
In family 1, EDA gene was investigated for mutation in two affected brothers and their mother. The same mutation of EDA gene was observed in two brothers and their asymptomatic carrier mother. Sequencing all the exons of EDA gene in family 1 revealed p.R156H (c.467G>A) mutation in exon 2 of EDA gene (Figure 1).
 missense mutation on <i>EDA</i> gene leading to X-linked disorder in two brothers; C, a heterozygous change in carrier mother")
A and B, EDA gene sequencing results show a (CGC>CAC) missense mutation on EDA gene leading to X-linked disorder in two brothers; C, a heterozygous change in carrier mother
In family 2, a 4-year-old boy affected with HED symptoms and his parents were investigated for mutation. After ruling out the presence of mutations in EDA, the EDAR gene remained as one of the main candidate genes linked to the disease to date. Sequence analysis of entire coding exons and the flanking intronic regions of the EDAR gene revealed one novel mutation c.1210G>A (p.A404T) in exon 12, where death domain is located (Figure 2).
 missense in the case of patient; C, control healthy samples were entirely homozygous for wild type allele.")
A, EDAR gene sequencing results show the same heterozygous GCA>ACA change in the parents; B, homozygous (GCA>ACA) missense in the case of patient; C, control healthy samples were entirely homozygous for wild type allele.
5. Discussion
EDA/EDAR pathway plays a crucial role in the development of many ectodermal organs in the embryonic stage. Mutations in genes encoding major components of the EDA/EDAR pathway disrupt normal ectodermal morphogenesis and lead to different forms of ED, which are indistinguishable with clinical signs examinations (6, 8).
In most HED cases, mutations in two main genes cause this disorder. The EDAR gene mutation results in autosomal dominant or recessive forms of HED, while mutations in EDA gene are responsible for X-linked HED, accounting for 90% of all HED cases (6).
EDA is a member of the tumor necrosis factor (TNF) superfamily, that can be cleaved in stalk region by furin to produce a secreted form to be ligand of EDAR (11). Therefore, furin cleavage sites in the stalk region are required for the EDA-mediated cell-to-cell signaling that regulates the ectodermal appendages development, as reported before (8).
In this study, we investigated the sequence analysis of EDA gene, the main gene involved in XHED, in two affected brothers and their carrier mother. The results in family 1 (with unrelated parents) revealed a previously reported missense mutation, G to A nucleotide substitution at codon 156 in exon 2 of EDA gene [p.R156H (c.467G>A)] at the furin cleavage site, which is a key consensus position site.
To date, more than 200 mutations have been reported for the EDA gene, most of which are missense, mostly seen in exons 1, 3, and 5 (1).
Regarding EDAR, which is the gene involved in autosomal form of HED, we identified an unreported missense mutation in the affected boy (homozygous) and his consanguineous parents (heterozygous) in family 2. EDAR is a type I transmembrane receptor protein with 448 amino acids. This protein belongs to TNF superfamily and consists of an extracellular region (in N terminal), a transmembrane region, and a death domain in the intracellular region (in C terminal). Previous studies showed that extracellular domain is necessary for binding to EDA ligand (8).
Our results showed that nucleotide substitution (c.1210G>A (p.A404T) - GCA>ACA) causes amino acid alanine (A) to change to threonine (T) in EDAR protein, where death domain is located.
Threonine is larger than alanine and more polarized with different tendencies for α helix formation. Not surprisingly, this change may affect the function of the protein. To date, over 50 mutations have been identified in this gene, of which 34 mutations are missense. Most of these mutations occur in exon 12, where death domain is located.
To confirm our findings, 120 unrelated healthy controls were selected and studied by Sanger sequencing to rule out genetic polymorphism. As mentioned earlier, the healthy control samples were entirely homozygous for wild type alleles so that the changes would not be a polymorphism.
For more reassurance, we used the mentioned bioinformatics tools to predict pathogenicity of novel detected mutation and their impact on EDAR functions. We used I-Mutant tool to predict the change of protein stability. I-Mutant tool predicted the effect of mutation on the protein as “decreased stability”. Sorting intolerant from tolerant (SIFT) has been used to human variants and is able to distinguish disease causing mutations from neutral polymorphisms. In our study, SIFT predicted the mutation as “damaging”. We also applied Polyphen-2, which predicts based on several parameters like phylogenetic, structural features, and sequence of protein. Polyphen-2 evaluated the potential of the alteration as “probably damaging”. In addition, Mutation taster was used to study the effects of missense mutations on disease-causing potential of sequence changes. Not surprisingly, the software predicted the impact of identified variant as “disease causing” (21).
Accordingly, all clinical evaluations, genetic findings, and computational analysis in this study supported the deleterious effects of both mutations on the EDA and EDAR genes. These findings can help to identify the exact genetic cause of these disorders in our population, and they might be useful for further studies and screening in different populations.
