


1. Introduction
Apert syndrome (OMIM #101200) is one of the severe forms of the craniosynostosis syndrome. The two frequent fibroblast growth factor receptor 2 (FGFR2) mutations (S252W and P253R) are responsible for 98% of cases. Autosomal dominant inheritance pattern has been reported, but most cases are due to a de novo mutation (1). It is classically characterized by midface hypoplasia, acrocephaly due to premature fusion of the coronal sutures and syndactyly. Complexity of symmetrical soft tissue and bony syndactyly in the hands and the feet (mitten hands) can differentiate it from other craniosynostosis syndromes. Other malformations in different organs can be associated with this syndrome particularly in central nervous system (CNS). Brain anomalies mainly involve midline structures and presenting as ventriculomegaly and corpus callosum agenesis/hypoplasia (1, 2). Temporal lobe abnormalities as overconvolution, and early clefts in Magnetic resonance imaging (MRI) of Apert cases were first described by Tokumaru et al. (3). Here we present a first case of 20 weeks fetus with temporal lobe abnormalities with a scalp cyst in fetal MRI.
2. Case Presentation
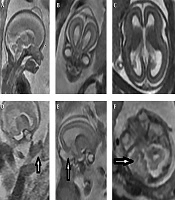
A 34-year-old woman (G1 P0) at 19 weeks and 3 days of gestational age with normal first trimester scan underwent second trimester anomaly scan. Ultrasound showed acrocephaly, mild ventriculomegaly (10.5 mm), hypertelorism, and mitten glove syndactyly of hands. Also a scalp cyst without calvarial bone defect was found. Fetal brain MRI on 1.5 T scanner confirmed 2D ultrasound findings. Also bilateral temporal lobe overconvolution and low set ears were detected by MRI. Corpus callosum was normal in ultrasound and MRI examination. These imaging features strongly suggested Apert or a related craniosynostosis syndrome (Figure 1). Amniotic fluid was obtained with an amniocentesis that revealed the heterozygous FGFR2 gene mutation c.758C > G (P253R) consistent with Apert syndrome. The parents chose termination of pregnancy. Postmortem examination of calvarium confirmed the existence of epidermoid cyst in scalp.

MRI findings of fetal brain at 20 weeks: A and B, Sagittal and coronal view show acrocephaly, scalp cyst, hypertelorism and normal corpus callosum; C, axial view shows mild ventriculomegaly; D, parasagittal view shows mitten glove syndactyly in hand; E and F: parasagittal and axial views show temporal lobe overconvolution.
3. Discussion
A few prenatal studies confirmed that temporal lobe abnormality is a very consistent feature in these fetuses when the brain is still relatively smooth (about 24 - 28 weeks) (1). The earliest reported temporal lobe malformation in prenatal life was about 23 weeks (4). Also temporal lobe abnormalities have been reported in other FGFR-related diseases, as Pfeiffer syndrome among other craniosynostosis syndromes and some skeletal dysplasias as thanatophoric dysplasia, achondroplasia, and hypochondroplasia. Overactivity of FGFR2 and 3 in the cortical hem has been proposed as a cause of temporal lobe overgrowth, and hippocampal dysplasia (1). Prenatal diagnosis of this syndrome by 2D ultrasound, though difficult, is usually based on skull shape abnormalities (as indirect sign of craniosynostosis), facial abnormalities, and syndactyly. MRI may have added value in evaluation of the brain anomalies, such as ventriculomegaly, agenesis of corpus callosum and especially temporal lobe abnormalities (4, 5) as in our case where temporal lobe overconvolution was only revealed by MRI. In normal fetus, temporal lobe sulci are only seen ≥ 26 weeks and presence of such clefting in early second trimester is clearly abnormal (1, 4). MRI and ultrasound are complementary tools for full evaluation of different aspects of a syndromic condition that is very important for proper genetic testing and counseling.
