



1. Introduction
Prader-Willi syndrome (PWS, MIM 176270) is an epigenetic disease caused by deletion or disruption of a gene or several genes in the 15q11-13 region. The incidence is estimated between 1/15 000 and 1/25 000 with an average mortality of 3% (1-3). The main clinical features include dysmorphic features, neonatal hypotonia, hypogonadism, severe hyperphagia, obesity, and cognitive impairment (4). We noted that about 1% PWS complicated with other hereditary diseases, including 47,XXY, DiGeorge syndrome, and mosaic variegated aneuploidy syndrome-1 among our over 400 PWS patients (unpublished data). Cerebellar ataxia, mental retardation and disequilibrium syndrome type 4 (CAMRQ4, MIM 615268) is a rare, autosomal recessive genetic disorder characterized by mental retardation, dysarthria, and truncal ataxia with or without quadrupedal gait. This syndrome is caused by the ATP8A2 gene (MIM 605870) mutation, which is located on chromosome 13q12 (5). AKAP10 gene (MIM 604694) located on chromosome 17p11 is related to another autosomal dominant cardiac conduction defect and cardiac susceptibility (MIM 115080). Here, we report a PWS infant with ATP8A2 and AKAP10 gene mutations, who presented multiple dysmorphic features and review correlative literatures.
2. Case Presentation
A 3-month-old boy presented to our unit because of developmental delay after birth. He was the gravida 4 and para 2 of the 38-year-old mother and 41-year-old father. His mother felt less fetal movement during pregnancy. He was born at 40+1 weeks’ gestation via cesarean delivery with normal hydramnios. His parents were not consanguineous marriage. He had a 16-year-old healthy brother.
Physical examination showed developmental delay and he still cannot raise his head. Characteristic facial features, including narrow bifrontal diameter, widely-spaced and almond-shaped eyes (often in mild upslanted position), blue sclera, full cheeks, small mouth, thin upper lip, downturned corners of the mouth, micrognathia (Figure 1A), high prominent forehead and low-set ear (Figure 1B) were noted. Moreover, he presented a peculiar position of the hands with the thumb constantly adducted over the index and middle finger (Figure 1C and D). Hypotonia, poor response, feeble cry, and empty scrotum was noted. Nystagmus and limb shaking were not noted.
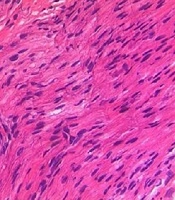
 Narrow bifrontal diameter, widely-spaced and almond-shaped eyes (often in mild upslanted position), blue sclera, full cheeks, small mouth, thin upper lip, downturned corners of the mouth, micrognathia; (B) High prominent forehead, low-set ear; (C-D) A peculiar position of the hands, with the thumb constantly adducted over the index and middle finger. Pathological images of muscle biopsy. (E) Small striated muscle tissue; (F) Large nerve fibers; (G) A small amount of compressed muscle fiber, dilated small blood vessels.")
Photos of current case. (A) Narrow bifrontal diameter, widely-spaced and almond-shaped eyes (often in mild upslanted position), blue sclera, full cheeks, small mouth, thin upper lip, downturned corners of the mouth, micrognathia; (B) High prominent forehead, low-set ear; (C-D) A peculiar position of the hands, with the thumb constantly adducted over the index and middle finger. Pathological images of muscle biopsy. (E) Small striated muscle tissue; (F) Large nerve fibers; (G) A small amount of compressed muscle fiber, dilated small blood vessels.
Brain ultrasonography showed a 0.67 cm × 0.31 cm hyperechoic zone under the left subependymal, which implied subependymal hemorrhage. Brain MRI showed small subependymal cysts which were about 0.3-0.5 cm in bilateral lateral ventricles. The Doppler echocardiography showed atrial septal defect (ASD, 0.2 cm) and patent ductus arteriosus (PDA, 0.1 cm). Ultrasonography noted the left and right testicle in the pelvic cavity near the bladder and dysplasia of hip joint (IIa). Muscle biopsy from right gastrocnemius muscle showed normal with small striated muscle tissue, dilated small blood vessels and large nerve fibers in a small piece of fibrous fatty vascular tissue (Figure 1E, F and G). Blood gas, electrolyte, liver and kidney function, GC/MS, thyroid function, auditory brainstem response, chest X-ray, ECG, ultrasonography for liver, gallbladder, pancreas, spleen, and kidney were all in the normal range or negative. ECG of his father and elder brother were all normal. He used nasogastric feeding for about 2 weeks because of poor feeding.
The neonatal department of our unit gave him whole-exome sequencing (WES) because of his abnormalities. The WES analyzed by our unit showed c.187C>G (p.P63A) in exon 2 originated from his father and c.2138T>C (p.I713T) in exon 23 originated from his mother, which were compound heterozygous variants of the ATP8A2 gene (Figure 2A and B). Bioinformatic analysis implied that both variants were missense mutations and not reported in ClinVar, HGMD, or LOVD databases. SIFT, Polyphen, and MutationTaster softwares implied that c.187C>G (p.P63A) variant might affect protein function at the general confidence level and regarded as low harm. As for the c.2138T>C (p.I713T) variant, the mutation might affect protein function at a lower confidence level and was regarded as high harm. In a word, the variants were assessed as uncertain significance. Moreover, a c.43delC heterozygous variant in exon 1 of the AKAP10 gene was also detected in our patient and his father (Figure 2C). Bioinformatic analysis implied that it was frameshift mutation and caused early termination in the 15th amino acid (p.L15fs). This mutation might affect protein function at a higher confidence level. It was not reported in ClinVar, HGMD, or LOVD databases, either. Therefore, the variant was assessed as potentially pathogenic.
 c.187C>G (p.P63A) in exon 2 originated from his father and c.2138T>C (p.I713T) in exon 23 originated from his mother were compound heterozygous variants of the <i>ATP8A2</i> gene; (C) A c.43delC heterozygous variant in exon 1 of the <i>AKAP10</i> gene originated from his father; (D) Normal copy numbers but abnormal methylation in the 15q11-13 region.")
Results of genetic testing. (A-B) c.187C>G (p.P63A) in exon 2 originated from his father and c.2138T>C (p.I713T) in exon 23 originated from his mother were compound heterozygous variants of the ATP8A2 gene; (C) A c.43delC heterozygous variant in exon 1 of the AKAP10 gene originated from his father; (D) Normal copy numbers but abnormal methylation in the 15q11-13 region.
Through the discussion of Multiple Disciplinary Team, the ATP8A2 and AKAP10 gene mutations could not explain his abnormalities. And his clinical manifestations were like PWS. Therefore, we gave him MS-MLPA test. The MS-MLPA text for Prader Willi/Angelman syndrome (MyGenostics Co., Ltd., Beijing, China) from peripheral blood revealed normal copy numbers but abnormal methylation in the 15q11-13 region, which implied nondeletion type PWS (Figure 2D).
When he was six months old, he came to our unit for follow-up. The Doppler echocardiography showed that ASD and PDA were self-healing. The left testicle was still in the pelvic cavity while the right testicle was noted in the inguina by ultrasonography. Developmental delay was especially obvious and he couldn’t sit.
3. Discussion
Our patient’s clinical manifestations and physical examinations were all consistent with PWS. And his genetic analysis supported this syndrome. Genetic evidence reveals that PWS is caused by functional loss of multiple paternally expressed genes (6). A gene or genes in the 15q11-13 region must be inherited from each parent for normal human development (7). If unexplained hypotonia and feeding difficulty appear during the neonatal period, physicians should suspect PWS. Infants who behave the typical craniofacial features of PWS should have genetic testing (8). The latest information on early diagnosis and management of PWS patients is significant for all physicians and will be useful in anticipating and managing or modifying complications related to this obesity-related disorder (9).
As our patient’s genetic testing showed, he had compound heterozygous variants of the ATP8A2 gene. The variants were not reported in any database. Whether he was complicated with other hereditary diseases remains to be discussed. The protein encoded by ATP8A2 is expressed in all brain regions, with the highest levels in the cerebellum, as well as in retina and testis (5, 10). The cerebellum is involved in bipedal locomotor activities and is especially important for movement control, balance, and locomotion (11, 12). Based on the above discussion, ATP8A2 might be crucial to the development of the central nervous system, and changes in this gene might result in severe neurological phenotypes. CAMRQ4 should be considered in patients who show ataxia, intellectual disability, hypotonia, microcephaly, choreoathetoid movements, ophthalmoplegia, and global developmental delay, even though brain MRI appears normal (13). Previous literatures described a total of 17 CAMRQ4 patients. Genetic analysis of them showed homozygous or compound heterozygous mutations of the ATP8A2 gene. Some couples of their parents were first-degree cousins. They all had early-onset, and their first features were hypotonia or developmental delay. In two girls specific time of symptom onset was described (14). One girl appeared abnormal involuntary movements in extremities and orofacial muscles, and horizontal nystagmus at the age of 2 years. When she was 6 years old, ophthalmoplegia and bilateral optic atrophy were observed. The other girl presented uncontrolled jerky movements, typical of chorea, in her face and four limbs at the age of 8 months. When she was 4.5 years old, she showed ophthalmoplegia and bilateral optic atrophy (14). Our patient had hypotonia and developmental delay, but these symptoms might be caused by PWS. In many cases, in the clinical course of these disorders when the patients grow older, often additional features appear. Given that our patient is still rather young, further observation is recommended.
Moreover, a c.43delC heterozygous mutation in exon 1 of the AKAP10 gene was also detected in our patient and his father. This mutation was not reported in any database, either. People with lower vagus nerve sensitivity have faster basal heart rate (HR), less variability of HR (HRV), and higher risk for sudden cardiac death (15). A single nucleotide polymorphism (SNP) resulting in an amino acid change from Ile to Val at position 646 in the AKAP10 gene is related to increased basal HR and decreased HRV, which shows that the Val variant has a negative effect on health (16, 17). AKAP10 mutant allele has a dominant interfering effect (16, 18). What’s more, the Val variant has greater atrioventricular node conduction than the Ile variant and is related to an increased risk of myocardial infarction (MI) (19). Our patient and his father both had normal ECG without clinical features related to heart, showing that the c.43delC heterozygous mutation of AKAP10 might be susceptible, and the effect of this mutation could appear as they become older. We should take the nonpathogenic possibility into account, so the patient needs further cardiac follow-up.
In conclusion, in patients with dysmorphic facial features, hypotonia, and developmental delay, PWS should be considered in the differential diagnosis. Moreover, other complicated hereditary diseases should be considered in patients with PWS.
