




1. Introduction
Wilson’s disease (WD), also known as hepatolenticular degeneration, is a hereditary autosomal recessive disorder caused by pathogenic variants within the ATP7B gene. WD is fatal when untreated, but with early detection and timely therapy the prognosis is quite good (1). In patients with WD, decreased biliary copper extraction leads to copper accumulation and injury, particularly in the liver, brain, and corneas. Patients with cirrhosis, neurological manifestations, and Kayser-Fleischer rings are easily diagnosed with WD, as these are the classic clinical manifestations, but Kayser-Fleischer rings are usually absent in children with WD (1). Furthermore, clinical manifestation of liver could be due to silent alteration in liver enzymes, chronic liver disease, and sub-fulminant as well as fulminant liver failure. Therefore, it is easy to misdiagnose WD in pediatric clinical practice. Fortunately, with the development of genetic testing, molecular analysis of the ATP7B gene is helpful for the early diagnosis of WD.
Hepatogenous diabetes (HD) is defined as a state of impaired glucose regulation caused by chronic liver disease (2). Although the association between liver cirrhosis and diabetes was first described in the 1960’s, it was scarcely studied for a long time (3). Recent studies have focused on the glucose metabolism in patients with chronic hepatitis C, and other chronic liver diseases (4-6). WD has been reported to be associated with elevated liver enzymes, hypoproteinemia, and coagulopathy (7). Here we report a child diagnosed as having WD with HD.
2. Case Presentation
A 4-year-old girl was admitted to Shanghai Children’s Medical Center in September 2018 with chief complaint of diarrhea for two months. The loose stool occurred five to six times per day without mucus or gross blood. She was not born from a consanguineous marriage. No medication use was reported at that time, and she was in good health before admission. Both her father and grandmother reported histories of mild hepatic dysfunction. A physical examination showed edema of the bilateral lower extremities. All the laboratory test including the coagulation tests are showed in Table 1. Meanwhile the characteristic chromatogram of warfarin was detected in peripheral blood and urine samples sent to Shanghai Poison Identification Center. To rule out food and drug poisoning, the patient’s medical history was examined further. It was revealed that the patient had regularly consumed Chinese Yinpian of reed rhizome for 3 years to relieve constipation. The instructions from this medicine indicated that one of its ingredients, esculin, has a chemical structure analogous to that of dicoumarin. Warfarin is a derivative of dicoumarin. Warfarin was no longer detected in her blood or urine after 12 days of cessation of the Yinpian consumption during hospital stay.
| Parameters | Values | Normal Range |
|---|---|---|
| WBC (× 109/L) | 17.25 | 4.0 - 15.0 |
| Hb (g/L) | 111 | 110 - 160 |
| Platelets (× 109/L) | 97 | 100 - 550 |
| PT (s) | 54.7 | 9 - 14 |
| APTT (s) | 87.3 | 28 - 45 |
| D-dmer (ug/ml) | 0.13 | < 0.3 |
| INR | 5.15 | 0.9 - 1.4 |
| ALT (U/L) | 328 | 13 - 39 |
| AST (U/L) | 338 | 15 - 46 |
| GGT (U/L) | 158 | 12 - 43 |
| ALB (g/L) | 23.1 | 35 - 50 |
| T-Bil (umol/L) | 69.5 | 0.30 - 22 |
| D-Bil (umol/L) | 67.8 | 0 - 19 |
Abbreviations: WBC, white blood cells; Hb, hemoglobin; PT, prothrombin time; APTT, activated partial thromboplastin time; INR, international normalalized ratio; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; ALB: albumin; T-Bil, total bilirubin; D-Bil, direct bilirubin.
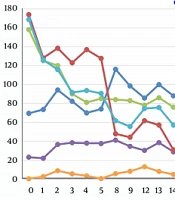
However, her liver function (Figure 1) and coagulation function (Figure 2) did not recover, and she developed lower extremity edema again, abdominal distension, and red urine on day 8 of admission. Laboratory tests showed progressively thrombocytopenia (platelets 29 × 109/L) and anemia (HGB 77 g/L). Viral hepatitis (A-E), Epstein-Barr virus, and cytomegalovirus infections were ruled out as causes of liver dysfunction. Autoantibody tests were negative.

The patient’s liver function test results during hospital stay

Coagulation test results during hospital stay
Morphological analysis of bone marrow revealed erythroid hyperplasia and morphologic evidence of a megakaryocyte mutation disorder. Both direct Coombs and indirect Coombs tests were negative. The patient was then diagnosed as having coagulation dysfunction with hemolytic anemia. Intravenous methylprednisolone (2 mg/kg*d) was administered for 23 days. The patient received transfusions of fresh frozen plasma, prothrombin proconvertin start power-factor B, plasma fibrinogen, albumin, glutathione, and VitK1. An infusion of platelets was also performed.
Abdominal contrast computed tomography (CT) showed liver parenchyma abnormality with diffuse small points of low-density, portal hypertension, a small amount of ascites.
Serum ceruloplasmin was 30 mg/L (normal, 200 - 400 mg/L), serum copper was 10.3 μmol/L (normal, 11.8 - 39.3 μmol/L), and urinary copper excretion was 156 μg/day (normal, < 40 μg/day). The suspected diagnosis was WD despite the absence of Kayser-Fleischer rings. With written consent from her parents, the genomic DNA of the patient and her parents was extracted from venous blood to detect mutations in ATP7B using QIAamp Blood DNA Mini Kit® (Qiagen GmbH, Hilden, Germany) to confirm the diagnosis.
On day 13 of hospitalization, the patient developed drowsiness with elevated blood ammonia levels (130 μmol/L), and physical examination showed right pupil dilation and a positive right Babinski sign. An emergent CT scan was performed to exclude intracranial haemorrhage. The patient was transferred to the PICU immediately for grade III encephalopathy. Mannitol was administered to reduce encephalic pressure, and arginine was transfused to lower blood ammonia levels. Cranial magnetic resonance imaging showed no structural abnormality. Both ADANTS13 activity and ADANTS13 antibody levels were normal. Emergency liver transplantation was recommended because of the life-threatening nature of the patient's illness, but this option was initially refused by her family.
On admission, urine glucose was 4+, fasting glucose was 2.6 mmol/L and postprandial blood glucose was 7.2 mmol/L. Over the following week, random blood sugar was between 7.3 mmol/L and 9.1 mmol/L, but the patient received no intervention. The abnormal blood glucose levels went unnoticed until day 24 of hospitalization, when the patient developed polydipsia, polyphagia, and polyuria with random blood glucose levels reaching as high as 12.1 - 18.9 mmol/L and as low as 1.8 - 3.3 mmol/L at morning measurements. Whole exome sequencing (WES) was performed to screen for other inherited metabolic disorders. One month after admission, the patient developed hypoglycemic convulsions when her blood sugar reached 0.6 mmol/L, and 25% glucose was transfused immediately. The patient’s parents then gave consent for liver transplantation. On day 40 of admission, the patient was transferred to Renji hospital and underwent successful orthotopic liver transplantation.
Sanger sequencing revealed compound heterozygous mutations in ATP7B, including c.2975C>T, p.Pro992Leu and c.3809A>G, p.Asn1270Ser (Figure 3A). The c.2975C>T mutation was also detected in the patient's father (Figure 3B), and the c.3809A>G mutation was detected in the patient’s mother (Figure 3C). No other pathogenic variants were detected by WES.
The patient was finally diagnosed as having WD. Her liver function, blood glucose levels, and coagulation test results returned to normal one month after the liver transplantation. Her stool was observed once a day, and the shape of the feces was type III according to the Bristol Stool Scale. Her abnormal glucose level in hospital was diagnosed as HD.
, patient’s father (B) and patient’s mother (C)")
ATP7B sanger sequencing of the patient (A), patient’s father (B) and patient’s mother (C)
3. Discussion
In the process of treating the patient, the importance of closely monitoring her blood glucose levels was not apparent, even with 4+ glycosuria on admission, until widely fluctuating blood sugar levels were observed several weeks later and the patient experienced hypoglycemic convulsions. Chronic liver disease can lead to a form of diabetes called HD. Different from classical type 2 diabetes, the HD is an acquired condition thought to be caused by impaired insulin clearance and pancreatic β cell dysfunction in cirrhotic patients (5). Liver dysfunction exerts a ‘toxic’ effect on pancreatic islets, playing a major pathophysiological role in the development of diabetes (8). In patients with chronic hepatitis C, the prevalence of HD increased with the degree of fibrosis, as did fasting glycemia (4). Recent studies revealed that HD was significantly associated with decreased survival rate and higher rate of liver complication. Nishida et al. (9) reported that 5-year cumulative survival rates of patients with cirrhosis with normal glucose tolerance, impaired glucose tolerance, and HD were 94.7, 68.8, and 56.6%, respectively. In another study, cirrhotic patients with subclinical abnormal glucose tolerance (SAGT) showed lower 5-year cumulative survival than those with normal glucose tolerance, and SAGT was independent predictors of death (10). Furthermore, HD is also associated with HE, variceal hemorrhage and spontaneous bacterial peritonitis (6-11). This patient’s glycosuria and elevated blood sugar were the result of abnormal glucose metabolism from liver cirrhosis in WD. Luckily, our patient’s blood sugar levels recovered one month after liver transplantation. Blood sugar monitoring should be performed in patients with WD, even when initial levels do not meet the diagnostic criteria for diabetes.
Numerous symptoms are observed in patients with WD. However, the symptom of chronic diarrhea, as observed in our patient, has rarely been documented as the initial symptom of WD. In our patient, the serum albumin level was < 2.8 g/dL, which was considered discordant with relatively moderate impairment of her liver function as evaluated by the Model For End-Stage Liver Disease (12). Chronic diarrhea could be a symptom of protein-losing enteropathy (PLE). PLE refers to an abnormally rapid loss of serum protein into the gut lumen (13). PLE may occur in patients with liver cirrhosis and can lead to worsening of hypoalbuminemia, ascites, and edema. Portal hypertension can lead to secondary intestinal lymphangiectasia and has been proposed as a cause of PLE in patients with liver cirrhosis. Stanley et al. (14) described a 41-year-old man with diarrhea, hypoalbuminemia, and cryptogenic cirrhosis with features of portal hypertension. PLE caused by portal hypertension was successfully treated with a transjugular intrahepatic portosystemic stent-shunt. Wong et al. (15) reported a case of a 32-year-old man with hepatitis B-related cirrhosis who presented with PLE that manifested as ascites and diarrhea. Portal hypertension was confirmed by Doppler ultrasound. Subsequently, the patient underwent living-related liver transplantation with complete resolution of PLE. The typical appearance of intestinal lymphangiectasia is white-tipped villi and white mucosa. Endoscopy is important for searching for intestinal lymphangiectasia in such cases. However, endoscopy was not performed in our patient because of thrombocytopenia and severe coagulopathy. The symptom of diarrhea was also relieved after liver transplantation in our case. Intestinal barrier dysfunction and bacterial translocation play an important role in the symptom of diarrhea (16).
Compound heterozygous mutations in the ATP7B gene were detected in the patient. The pathogenic c.2975C>T variant is common in Chinese patients with WD (17), while the pathogenic c.3809A>G variant is observed less frequently, it has but been reported to cause WD(18). It is reported that the patients with c.2975C>T or c.3809A>G often present WD features before 12 years of age (17). Recently, WES has allowed for the analysis of all exons of all protein-coding genes and is considered a useful tool for detecting disease allelic variants underlying Mendelian disorders and rare disease-related genomic alterations (19). In this case, WES was also applied but no other pathogenic variants were detected.
In conclusion, blood glucose levels must be carefully monitored in Wilson’s disease. Furthermore, this case report demonstrates that genetic sequencing provides an accurate and minimally invasive diagnostic tool for WD.
