




1. Introduction
Renpenning et al. (1) published an article in 1962 with 20 male patients from three generations with mental retardation (MR), which were associated with microcephaly, prominent ears, cataracts, congenital ocular coloboma, and seizures. Turner et al. (2), from a group of 148 males with intellectual disability (ID), analyzed 17 who were considered to have that same syndrome because of the same pattern of inheritance. Scientists mapped the locus to Xp11.2-p11.4 and suggested that the syndrome with this mutation should be called Renpenning syndrome (3). In five of 29 families with X-linked mental retardation (XLMR), the deletion/duplication of an AG dinucleotide on proximal Xp in the polyglutamine tract-binding protein 1 (PQBP1) gene was identified, causing frameshift in the fourth coding exon (4). Stevenson et al. (5) identified a 4-bp deletion in exon 4 of the PQBP1 gene denoted by the mutation as 459-462delAGAG. It was then proposed that all mutations in the PQBP1 gene whose clinical manifestations over time received different names in the literature were unified under the same name: Renpenning syndrome (5). Also, this mutation in the PQBP1 gene (the fourth coding exon) has been described by other authors (4-7).
Authors showed two male relatives who had a deletion in PQBP1 gene, and one of them had seizures, which was rarely described in previous cases (8). The first case related to an 18-year-old female was also reported (9). Streaky hypopigmentation of the skin was an additional finding for this syndrome.
The data indicated that the life expectancy of these patients was not significantly shortened. A total of four boys died due to the primary series in childhood (one died during febrile convulsions) (1). Follow-up of this family by other authors showed that these men reached middle adulthood and old age (3, 10).
2. Case Presentation
In this case report, we presented an 11-year-old male who was born of a full-term and uncomplicated pregnancy and experienced mild developmental delay, especially in speech skill. He began sitting at the age of 8 months, crawling at 12 months, and walking at 15 months. The first words came at the age of 12 months, but the speech progressed slowly toward a complex language.
Psychological assessment at the age of seven years indicated intellectual functioning at the level of mild ID (MID), with a statistically higher score on the verbal than nonverbal scale. The behavior was dominated by low frustration tolerance and hyperactivity. Currently, the boy attends a primary school in a special individual education program and is well-suited in the peer group.
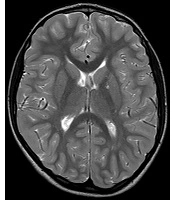
The following morphological abnormalities were detected: microcephaly (with head measurement at birth 30 cm, < 3rd centile), wide nasal bridge, midfacial and maxillary hypoplasia, short philtrum, low-set ears, low hanging columella, high palate, and narrow face. Neurological examination showed on upper and lower extremities hypotonia with joint hypermobility. The rest of neurological examination was normal. A systolic murmur of intensity 2/6 was present in the cardiac findings. Echocardiography showed chordae tendineae abnormalities in the left ventricle, which was not clinically significance. The patient had his first seizure at the age of seven, and he experienced four nocturnal seizures by the age of nine. Seizure began with nausea, an urge to vomit, a blank stare, an absence of response to calls. The attacks lasted up to three minutes. Electroencephalography (EEG) was performed and showed focal epileptiform changes left anteriorly (Figure 1). Lamotrigine at a dose of 5 mg/kg was prescribed. Brain magnetic resonance imaging (MRI) showed asymmetric cerebellar hemispheres (mild right cerebellar hemisphere hypoplasia) (Figures 2 and 3). There were no other pathological changes on brain MRI (Figure 4). During the next 18 months, he was without seizure, and EEG performed on two occasions was without specific discharge. At the age of 10, in one month, he had exacerbation with five seizures without known triggers with different semiology to a certain extent: head and sideways view, body cramping, jaw clenched, no contact with boy, sweated. The attacks lasted for 1 - 3 minutes. Description corresponds to a seizure with onset in sleep with evolution in bilateral motor seizure. Further monotherapy with increased dose of lamotrigin showed benefit, with only one repeated seizure at the age of 11. At that age, his weight was 26 kg, and height was 110 cm (short stature).

EEG showed focal epileptiform changes left anteriorly

Axial T2W MRI showed a mild right cerebellar hemisphere hypoplasia

Sagittal MRI showed a mild cerebellar hemisphere hypoplasia

Absence of other pathological changes on brain MRI
A karyotype analysis was performed and showed the male karyotype 46, XY. After that, frameshift mutation in polar reach domain (PRD) of the PQBP1 gene (c.459-462 delAGAG) was detected by exome sequencing. The mother of our patient was a heterozygote for the same mutation.
3. Discussion
In this study, we showed the first case from Serbia of a boy with MID, microcephaly, dysmorphic face, short stature, and seizures who found a mutation in the PRD of the PQBP1 gene. Brain MRI showed mild unilateral cerebellar hemisphere hypoplasia.
ID, microcephaly, short stature, and small testicles constitute a tetrad in a typical patient with a mutation in the PQBP1 gene. The craniofacial manifestations (central balding, upslanted palpebral fissures, short philtrum, everted lower lip, and narrow tall face) may thus be helpful, but probably not sufficient for diagnosis (1, 3, 5). The cardiac malformations are common in this syndrome, and chordae tendineae abnormalities in the left ventricle were seen in our patient. The authors considered that patients with genetic mutation, as our patient, had the mildest clinical presentation (5).
Three subjects with Renpenning syndrome had epilepsy (1), and one of them had seizures with generalized onset during childhood (3). Epilepsy in children with mutation in the PQBP1 gene appears to be the exception rather than the rule since Stevenson et al. (5) found seizures reported in 11% of affected males. Another patient who had epilepsy had the same mutation as our patient and associated microphthalmia (8). In our patient, epilepsy began at the age of seven. The attacks had a focal onset in sleep with evolution into a bilateral motor attack. Epilepsy was relatively well controlled with monotherapy.
Thin posterior part of the corpus callosum (7) and nodular gray matter heterotopia [6] had pathological findings on brain MRI associated with a mutation in the PQBP1 gene. Germanaud et al. (7) showed that the cerebellum was reduced in proportion to the whole brain, mainly due to the reduction of the dimensions of the vermis; this finding was partially supported by our patient’s MRI findings of asymmetric cerebellar hemispheres.
The mutations in PRD domain of the PQBP1 gene detected in our patient were considered to be the most important for clinical manifestations of Renpenning syndrome (11). This is considered one of the main mechanisms by which this mutation leads to the microcephaly (12).
The association between mutations of certain genes (CASK, OFD1, MID1, AP1S2, ARX) on the Xp chromosome with mild or global cerebellar hypoplasia on neuroimaging as part of very rare syndromes (CASK, Oral-facial-digital type I/X-linked Joubert, etc.) has been demonstrated (13). The CASK gene whose mutation causes CASK syndrome is the closest to the PQBP1 gene on the X chromosome and is at the Xp11-4 position. The phenotypes associated with CASK vary from MID to severe cognitive impairment associated with cerebellar and pontine hypoplasia and cortical development abnormalities.
Beside the connection with microcephaly, the role of PQBP1 gene can be very significant in the pathogenesis of neurodegenerative diseases such as Huntington’s disease (14) and spinocerebellar ataxia 1 (15), which puts this gene at the heart of the “two-hit hypothesis” of neurodegeneration. The essence of the “two-hit hypothesis” is that misfolded protein (produced during neurodegenerative disease) interacts before aggregation with PQBP1 gene mutant protein, which triggers cell dysfunction and cell death (11). Thus, asymmetric cerebellar hemispheres on brain MRI in our patient (from one side) and the link of this mutation (PRD of the PQBP1 gene) and the neurodegenerative diseases (from the other side) may be related.
3.1. Conclusions
Our patient had classical clinical manifestations for Renpenning syndrome (microcephaly, ID, and short stature), which have been proven to be the most common frameshift mutation in the PRD of the PQBP1 gene. To the best of our knowledge, this is the first patient with Renpenning syndrome with asymmetric cerebellar hemispheres (mild right cerebellar hemisphere hypoplasia). We do not know whether cerebellar hypoplasia is part of syndrome or just a sign of comorbidity and whether the occurrence of epilepsy in Renpenning syndrome with cerebellar hypoplasia is more likely. Further radiological, neurophysiological, and clinical follow-ups on Renpenning syndrome cases with a larger sample size are necessary.
