




1. Introduction
Neonatal diabetes mellitus (NDM) is defined as the onset of diabetes mellitus within the first six months of life (1), which is usually not autoimmune based. A genetic cause can be found for nearly more than half of the NDM patients (1). Mutations in ABCC8 and KCNJ11 are the most known common causes of NDM. Pancreatic and cerebellar agenesis are rare causes of neonatal diabetes. The pancreas-specific transcription factor 1A (PTF1A) gene encodes a transcription factor that plays a critical role in the early pancreas and cerebellar development (2). Mutation in PTF1A can impair this process and result in NDM. These patients show phenotypic variability, such as different ages for diabetes onset or involvement of cerebellum (2, 3).
Biallelic truncating variants in PTF1A have been reported in patients with pancreatic and cerebellar agenesis, whereas variants in the distal pancreatic-specific enhancer of the gene may cause isolated pancreatic agenesis (2). Here, we investigated the clinical features and genetic causes of NDM. We also reviewed clinical manifestations of previously reported cases of NDM caused by PTF1A mutation.
2. Case Presentation
The female neonate from a consanguineous Iranian family was presented during the first month of life with permanent neonatal diabetes. The pregnancy was uneventful. Fetal ultrasonography was normal. She was born with a gestational age of 39 weeks by cesarean section due to an abnormal non-stress test, suspicious of fetal distress. At birth, she passed meconium, and her Apgar score was standard. Her parents were first cousins; her grandmother was a known case of adulthood type 2 diabetes mellitus.
At birth, the body measurements were as follows: birth weight 1,800 g, length 43 cm, and head circumference 33 cm. Physical examination was normal, with no skeletal deformity or facial dysmorphism. She was admitted due to intrauterine growth retardation (IUGR). The hospital course was complicated by pneumonia and sepsis, and she was admitted to Neonatal Intensive Care Unit (NICU) for 21 days. High blood sugar levels (400 mg/dL) were recorded since the 17th day of life but were thought to be "stress hyperglycemia". She received a few doses of regular insulin and was discharged at 21 days of age in good condition, without the maintenance of the insulin.
She was admitted again on the 53rd day of age because of chronic diarrhea and direct hyperbilirubinemia. Total and direct bilirubin levels were 9.7 mg/dL and 3 mg/dL, respectively. During this admission, the patient had repeated episodes of hyperglycemia. Therefore, regular and NPH insulins were started. Laboratory investigations showed fat malabsorption. Stool exam showed increased excretion of neutral fat (> 60 drops of neutral fat and > 100 drops of fatty acid in each microscopic field). Fecal elastase level was lower than 50 mg/gr (normal 200 - 500 mg/gr). Ophthalmoscopic exam and thyroid and renal function tests were normal. Liver enzymes were mildly elevated (ALT 72 u/L, AST 64 u/L), but this increase was transient, and the enzyme levels returned to the normal range. She had hypoproteinemia and hypoalbuminemia, with a total protein of 3 gr% and albumin of 2.5 gr%. Other laboratory tests were normal, including serum ammonia, PT and INR, 25-hydroxyvitamin D, amylase, and lipase.
Ultrasonographic study of the abdomen showed a normal-sized pancreas (length 50 mm and thickness 5 - 8 mm). magnetic resonance imaging (MRI) of the brain was completely normal without cerebellar involvement. Anti-islet cell antibody was negative, anti-glutamic acid decarboxylase (GAD) antibody was borderline (13.6 units/mL, normal range < 10 units/mL) and anti-insulin antibody was elevated (60 unit/mL, normal < 10 units/mL). Antibody measurements were performed a few months after starting insulin therapy, and high anti-insulin antibody levels can be secondary to exogenous insulin. The patient was discharged home in good general condition.
Treatment continued with NPH and regular insulin and exogenous pancreatic enzymes (creon, Solvay Healthcare, Hannover, Germany). At 18 months of age, the insulin regimen was switched to analog insulin, Levemir, and Apidra, with a dose of about 1unit/kg/day, creon was continued.
During the last visit at 23 months, the patient’s body measurements were as follows: weight 11.6 kg (38th percentile), length 82.7cm (22nd percentile), with normal development and normal neurological and general physical examination.
2.1. Genetic Analysis
Genomic DNA was isolated from the peripheral blood of the patient. Sequencing the coding regions of ABCC8, KCNJ11, INS, and EIF2AK3 genes did not identify a pathogenic variant. PTF1A gene sequencing covered the coding exons and distal enhancer (∼400 bp sequence located 25 kb downstream the gene) using Sanger sequencing, as previously described (4). A single homozygous nucleotide variant (Chr10, g.23508441T > G) affects a highly conserved nucleotide within a distal enhancer of the PTF1A gene. Variants in this enhancer region mutation of a new distal developmental enhancer of PTF1A are a common cause of isolated pancreatic agenesis in humans (4). Specifically, two adjacent variants, g.23508437A > G and 23508446A > C have been previously identified in patients with isolated pancreatic agenesis (4). Tutak et al. reported an infant with neonatal diabetes and cerebellar agenesis who died at 42 days of age, but his mutation was not studied. Both of his parents were heterozygous for frameshift mutation (G240fsX276) in the PTF1A gene (5).
3. Discussion
We report a case of permanent neonatal diabetes, resulting from a homozygous PTF1A variant. Initial documented high blood glucose level was in the 1st month of life, which persisted until the last visit at 23 months old. She had pancreatic exocrine insufficiency requiring exogenous enzyme replacement; however, the pancreas was normal in ultrasonography. She did not have any neurological manifestation, and the brain MRI was normal.
3.1. Review of Previously Reported Cases
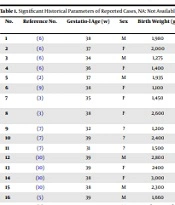
We reviewed the clinical features of previously 18 published cases (including our case) of NDM with a PTF1A variant. Table 1 shows the historical parameters of reported cases, and Table 2 compares their clinical and Paraclinical findings. Most cases were from the Middle East. Six cases (33%) were products of preterm deliveries (gestational age < 37 weeks) (3, 6-8), and the lowest gestational age was 31 weeks (7). The preterm birth did not affect clinical manifestations or prognosis. All except three of these cases had low birth weight (over 80%), probably because insulin has a significant role in fetal growth. The onset of hyperglycemia and initiation of insulin therapy in 14 patients were within the first month of life (78%), which can be due to the effect of PTF1A variants on the fetal development of the pancreas. The presentation of hyperglycemia in one patient was as late as mid-childhood (9.5 years). A significant number of patients (44%) had failed to thrive (FTT), which was apparently due to malabsorption and not diabetes or neurological involvement. The growth of patients without malabsorption was within normal ranges (3, 6). The development of 10 patients (for which data was available) was normal. Five patients (28%) with high glucose levels immediately after birth died in early infancy (5, 6, 8, 9), so no clear correlation was identified between the age of onset and death. Little information was available regarding brain development. In one of the previously reported cases (6) and our patient had a normal brain MRI. However, semi-lobar holoprosencephaly was reported in one patient (9).
| No. | Reference No. | Gestatio-l Age (w) | Sex | Birth Weight (g) | Age at Presentation | Consanguinity | Development | Ethnic or Country of Origin |
|---|---|---|---|---|---|---|---|---|
| 1 | (6) | 38 | M | 1,980 | 1 day | Yes | Normal | Saudi Arabia |
| 2 | (6) | 37 | F | 2,000 | 1 day | Yes | Normal | Saudi Arabia |
| 3 | (6) | 34 | M | 1,275 | 1 day | Yes | Normal | Kuwait |
| 4 | (6) | 36 | F | 1,400 | 8 days | Yes | Normal | Kuwait |
| 5 | (2) | 37 | M | 1,935 | 7 days | No | - | European |
| 6 | (9) | 38 | F | 1,100 | 1 day | No | - | European |
| 7 | (3) | 35 | F | 1,450 | 10 days | Yes | Normal | Half-Turkish Half-Kurdish |
| 8 | (3) | 38 | F | 2,600 | 9 years | Yes | Normal | Half-Turkish; Half-Kurdish |
| 9 | (7) | 32 | ? | 1,200 | 3 weeks | Yes | - | Turkish |
| 10 | (7) | 39 | ? | 2,400 | 10 weeks | Yes | - | Turkish |
| 11 | (7) | 31 | ? | 1,500 | 1 week | Yes | Delayed | Turkish |
| 12 | (10) | 39 | M | 2,800 | 4 weeks | Yes | Normal | - |
| 13 | (10) | 39 | F | 2400 | 3 weeks | Yes | Normal | - |
| 14 | (10) | 38 | F | 3,000 | 78 weeks | Yes | Normal | - |
| 15 | (10) | 38 | M | 2,300 | 2 weeks | Yes | Normal | - |
| 16 | (5) | 39 | M | 1,660 | 2 days | Yes | Delayed | Turkish |
| 17 | (8) | 36 | M | < 3rd percentile | 2 months | Yes | Delayed | Saudi Arabia |
| 18 | This paper | 39 | F | 1,800 | 17 days | Yes | Normal | Iranian |
Significant Historical Parameters of Reported Cases
| No. | Reference No. | Facial Dysmorphism | Failure to Thrive | Malabsorption | Neurological Involvement | Imaging of Pancreas | Imaging of Brain | Outcome | Mutation |
|---|---|---|---|---|---|---|---|---|---|
| 1 | (6) | - | Yes | Yes | No | Not identified pancreas in ultrasonography | Not performed brain MRI | Alive | c.571C > A |
| 2 | (6) | - | No | No | No | Not identified pancreas in ultrasonography | Not performed brain MRI | Alive | c.571C > A |
| 3 | (6) | - | Yes | Yes | No | - | Not performed brain MRI | Died at 12 weeks | |
| 4 | (6) | - | Yes | Yes | No | - | Normal brain MRI | Alive | |
| 5 | (2) | - | Yes | Yes | - | Pancreas could not be identified in ultrasonography | - | Alive | Compound hetero g.23508442A > G and… |
| 6 | (9) | Yes | Yes | Yes | Yes | Absence of pancreas and gallbladder in abdomen | Brain MRI semilobar holoprosencephaly | Died at 12 weeks | |
| 7 | (3) | - | Yes | Yes | Yes | Severe pancreatic hypoplasia of head, tail and body were nearly undetectable | - | Alive | Homozygous for g.23508437A > G |
| 8 | (3) | - | No | No | No | Pancreatic hypoplasia | Normal brain MRI | Alive | Homozygous for g.23508437A > G |
| 9 | (7) | - | - | - | - | Pancreatic hypoplasia | - | Alive | Homozygous for g.23508365A > G |
| 10 | (7) | - | - | - | - | Pancreatic hypoplasia | - | Alive | Homozygous for g.23508437A > G |
| 11 | (7) | - | - | - | Yes | Pancreatic hypoplasia | - | Alive | Homozygous for g.23508437A > G |
| 12 | (10) | - | - | Yes | - | Pancreatic agenesis | - | - | |
| 13 | (10) | - | - | Yes | - | Pancreatic agenesis | - | - | |
| 14 | (10) | - | - | Yes | - | Pancreatic hypoplasia | - | - | |
| 15 | (10) | - | - | Yes | - | Pancreatic agenesis | - | - | |
| 16 | (5) | Yes | Yes | - | Yes | - | Absence of cerebellum | Died at 42 days | Not checked |
| 17 | (8) | Yes | Yes | - | Yes | Absence of pancreas | Cerebellar agenesis | 4 months | c.437-460del |
| 18 | This paper | No | No | Yes | No | Normal | Normal | Alive | g.23508441T > G |
Significant Clinical and Paraclinical Findings in Reported Cases
In 15 (out of 18) cases, pancreas imaging was performed. The pancreas was not detected (2, 6, 8-10) in eight cases, the pancreas was hypoplastic in six subjects, and the pancreas was normal in our patient, whereas the endocrine and exocrine functions were abnormal. These results may suggest that PTF1A variants can cause pancreas function abnormality, even without a significant structural defect.
Based on our review, PNDM is a genetically heterogeneous disorder due to mutations in 23 different genes described to date: KCNJ11, ABCC8, FOXP3, GCK, PDX1, PTF1A, EIF2AK3, SLC2A2, GATA6, GATA4, SLC19A2, WFS1, NEUROD1, NEUROG3, RFX6, LRBA, NKX2-2, MNX1, IER3IP1, INS, STAT3, GLIS3, and HNF1B. However, mutations in the genes encoding the ATP-sensitive potassium channel (KATP) subunits, KCNJ11 (Kir6.2), ABCC8 sulphonylurea receptor 1 (SUR1), and INS (insulin) compromise insulin secretion by affecting the mechanisms involved in insulin secretion. Also, previous functional analyses showed that a homozygous mutation could distrust enhancer activity and is likely to decrease in PTF1A expression during pancreatic development. This result confirms a diagnosis of neonatal diabetes and exocrine pancreatic insufficiency due to a recessive PTF1A mutation (11, 12). Therefore, we evaluated sequencing the coding regions of ABCC8, KCNJ11, INS, and EIF2AK3 genes and also the PTF1A gene in our study. We report a case of permanent neonatal diabetes, associated with a homozygous PTF1A variant.
3.2. Limitations
There were some limitations in our study. Due to financial and time constraints, we evaluated sequencing of only coding regions of the other four genes (ABCC8, KCNJ11, INS, EIF2AK3), while other genes such as PNDM: PDX1, RFX6, GATA4, GATA6, GLIS3, NEUROD1, PAX6, MNX1, NKX2-2, GCK, etc. that might have been related to permanent neonatal diabetes were not screened. The screening of them is suggested in another study.
3.3. Conclusions
Our study showed that there is an association between the identified PTF1A gene variant and permanent neonatal diabetes. Since most previously reported patients had IUGR, and imaging evidence of pancreatic agenesis or hypoplasia, NDM due to PTF1A variants should be considered in the differential diagnosis of neonatal diabetes (especially with the onset in the first month of life). We suggest that pancreatic imaging and evaluation of exocrine pancreas function can help early confirm the diagnosis in these patients. Our results expand the clinical phenotype associated with PTF1A enhancer mutations and highlight the importance of genetic testing and clinical follow-up studies to characterize rare genetic subtypes of diabetes. These results can be applied in screening and genetic counseling, especially in Middle Eastern populations.
