


1. Introduction
Mitochondrial diseases are among the most prevalent inborn metabolic disorders caused by disturbances in the oxidative phosphorylation (OXPHOS) system (1). They can manifest at different ages, mimic several disorders, and be diagnosed after ruling out other clinical conditions, confirmed by genetic testing (2). Leigh Syndrome (LS) encompasses a spectrum of mitochondrial diseases initially described in 1951 by Leigh as a necrotizing encephalopathy with bilateral necrotic lesions in basal ganglia, thalamus, and brain stem (3). Currently, LS is designated for all progressive encephalomyopathies with symmetric necrotizing lesions, and its classic form is also known as infantile necrotizing encephalopathy. Leigh syndrome classically begins in the first year of life with progressive neurological deterioration and usually is accompanied by lactate acidosis resulting from decompensation in the oxidation chain. Neurological symptoms vary and involve dystonia, spasticity, epilepsy, chorea-like movements, and blindness (4). Leigh syndrome is usually inherited as an autosomal recessive trait; however, it has been observed with mitochondrial DNA and X-linked inheritance, as well. Defects in more than 70 genes have been linked to the pathogenesis of LS, mainly through disruption of cell energy production (4, 5).
Mitochondrial respiratory chain complex I is the largest complex of the respiratory chain with 45 separate polypeptides in mammalian cells, which contains three functional modules: (1) the Q-module (ubiquinone reduction); (2) the N-module (NADH dehydrogenase); and (3) the P-module (proton translocation). NDUFA9, a component of the Q-module, is part of an intricate process essential for complex I assembly or stability (6-8).
NDUFA9 (OMIM#603834) is one of the latest detected genes responsible for LS (9). It comprises 11 exons and is located on chromosome 12 (10). So far, only two cases of LS with pathogenic missense variations in NDUFA9 have been described, both affecting a highly conserved C-terminal part of the coded protein, affecting the complex I assembly (9, 11). We present another patient with a Leigh-like phenotype with a novel missense variation in NDUFA9. The study was approved by the Research and Ethics Board of Qazvin University of Medical Sciences, and written informed consent was obtained from the parents.
2. Case Presentation
The patient was a male neonate and the first offspring of a consanguineous (first cousins) Iranian couple of Turkish ethnicity, born at full-term through natural vaginal delivery and without any past medical history or antenatal insults. His birth weight was 2,900 g, and the Occipitofrontal circumference was 35 cm. For two days following his birth, he had been under observation for icterus and a diagnosis of transient tachypnea of neonates (TTN) but was discharged in good condition. On the 17th day of birth, the patient was again hospitalized with poor sucking and tachypnea complaints. Due to the observed episodes of cyanosis, he went through different work-ups for sepsis, cardiovascular and pulmonary abnormalities, and seizures. Electroencephalogram was compatible with seizure activity. Ultimately, he was discharged on levetiracetam and phenobarbital with the impression of a seizure. He still experienced the same cyanotic episodes and suspicious jerks within the following month.
He was re-admitted to the emergency department with the respiratory arrest at 58-days old. The patient was successfully resuscitated but became ventilator-dependent due to a low level of consciousness and difficulty breathing. In his metabolic work-ups, high plasma levels of ammonia (2.4 mg/L) and lactate (96 mg/dL) were detected. Tandem mass spectrometry (MS/MS) showed mild elevations in branched-chain amino acids (BCAA), leucine, and isoleucine, along with methionine, valine, Malonyl carnitine/3-Hydroxybutyrylcarnitine, and methylmalonyl carnitine/3-OH isovaleryl carnitine. These findings were confirmed by serum amino acid analysis with high-performance liquid chromatography (HPLC), showing serine, methionine, valine, leucine, and isoleucine abnormalities. Other supplementary paraclinical studies included lumbar puncture and metabolic analysis of cerebrospinal fluid, showing elevated lactate levels. Moreover, he underwent a computed tomography (CT) scan demonstrating hydrocephalus, bilateral ventriculomegaly (maximum diameter = 13 mm in the left lateral ventricle), and diffusely diminished white matter density. He had no cardiovascular or respiratory system abnormalities, and other routine laboratory data were in normal ranges.
Considering the presentation and paraclinical findings, he was initially diagnosed with Maple Syrup Urine Disease (MSUD), and thus a specific treatment regimen was initiated; nevertheless, his condition deteriorated over time. Gradually, he developed nystagmus and spastic paraplegia and failed to reach his developmental milestones. As he was unresponsive to the optimal management of MSUD, other syndromic diagnoses were considered. Moreover, his family was consulted for genetic testing, and subsequently, a whole-exome sequencing (WES) was performed; however, the patient expired when he was four months old due to respiratory insufficiency. His WES demonstrated a novel biallelic, autosomal recessive missense variation (c.1069C>G, p.Arg357Gly) in the NDUFA9 gene.
2.1. Genetic Testing
DNA was extracted from the peripheral blood leukocytes of the proband using a commercial kit (high-pure PCR Template Preparation, Roche). We then performed WES on peripheral blood DNA. In brief, DNA was captured with the Agilent SureSelect v.7 exome capture oligonucleotide library and sequenced paired-end 100 bp reads on Illumina HiSeq 2000 by standard Illumina protocol. Bioinformatics analysis of the resulting reads was performed using BWA aligner, GATK, and ANNOVAR. The reads were aligned to the reference genome build GRCh37/hg19 using the BWA aligner tool, and variants were identified using GATK and annotated using ANNOVAR software. More than 110 K variants were annotated and filtered based on their frequency, CADD score, functional consequences, mode of inheritance, and the related clinical phenotype. This analysis led to identifying a rare novel homozygous missense variation in the NDUFA9 gene, changing Arginine 357 to Glycine (NM_005002.5: exon 11: c.1069C>G, p.Arg357Gly) in the proband. Sanger validation of the variant confirmed that the proband was homozygous, and both parents were heterozygous at the variation position (Figure 1). Detailed computational analysis of p.Arg357Gly variation using prediction methods (PolyPhen-2, SIFT, VariationTaster) predicted it as a Variant of Unknown Significance (VUS) (Table 1). The frequency of the variant in question is extremely rare (zero) in all population databases, including gnomeAD (Aggregated), TOPMed Bravo, ClinVar, GME Variome, ExAC, 1000 Genomes, ESP 6500, 4.7 KJPN, and Genome Asia. The local NGS database (http://www.iranome.ir/), currently consisting of 1,406 WES data from healthy controls, was investigated, and no samples exhibited the identified novel variation in NDUFA9.
, heterozygous and homozygous mutant.")
A, Family pedigree of the patient; B, NDUFA9 structure and variations; C, The highly conserved state of the variant amino acid across the evolution of species; D, Electropherograms from Sanger confirmation in family members showing NDUFA9 (c.1069C>G, p.Arg357Gly), heterozygous and homozygous mutant.
| Computational Tools | Prediction | Score | Rank score |
|---|---|---|---|
| BayesDel addAF | Damaging | 0.2578 | 0.7932 |
| BayesDel noAF | Damaging | 0.1325 | 0.7905 |
| DEOGEN2 | Tolerated | 0.3327 | 0.7026 |
| EIGEN | Pathogenic | 0.7797 | 0.8482 |
| FATHMM | Damaging | -1.67, -1.37 | 0.8281 |
| MutPred | Damaging | 0.73 | 0.8637 |
| Mutation assessor | Medium | 3.235 | 0.8961 |
| MutationTaster | Disease Causing | 1 | 0.81 |
| PROVEAN | Damaging | -3.61, -5.63 | 0.8684 |
| SIFT | Damaging | 0, 0.002 | 0.9125 |
Computational Analysis and Pathogenicity Prediction of NDUFA9, c.1069C>G Variation
3. Discussion
We presented an infant with a fatal progressive neurodegenerative disorder, likely triggered by a genetically determined variation (c.1069C>G, p.Arg357Gly) in exon 11 of NDUFA9. This variation, which has not been reported before, is predicted to be a VUS based on the American College of Medical Genetics (ACMG) classification and computational analysis. NDUFA9 consists of 11 exons, coding for 377 amino acids. Two different NDUFA9 missense variations have been reported in unrelated families from various origins (6, 8). Pathogenic variations affecting this gene are believed to cause LS; however, at first, the case was mistaken for a disorder of BCAA metabolism, MSUD, since the absolute levels of BCAA were high in both HPLC and MS/MS. Moreover, the patient showed some signs and symptoms, including lethargy, metabolic acidosis, and spasticity, usually seen in MSUD, LS, and many other diseases caused by inborn errors of metabolism in infancy. Previous studies have shown that disorders of the mitochondrial respiratory chain can elevate BCAA (12). Furthermore, early-onset type 3 MSUD, caused by dihydrolipoamide dehydrogenase (DLD) deficiency, can present with manifestations similar to LS (13). A differential diagnosis of mitochondrial disorders should be considered for MSUD, especially in patients unresponsive to MSUD management.
Two cases have been reported with variations in NDUFA9 causing LS. The first case was a Kurdish boy born to a consanguineous family with a normal perinatal period and birth. In the first few days of birth, he presented with sensorineural hearing loss, retinitis pigmentosa, lactic acidosis, LS-characteristic lesions in brain magnetic resonance imaging (MRI), and diffuse loss of white matter and brainstem volume. His electroencephalogram was compatible with seizure, and he had choreo-dystonic movements in all four limbs. Laboratory tests returned high levels of lactate and slightly elevated alanine levels. He experienced apnea spells, which were hypothesized to be due to brainstem involvement. The patient died at one month of age due to respiratory failure. Homozygosity mapping followed by targeted next-generation sequencing (NGS) revealed a missense likely pathogenic variation in exon 10 of NDUFA9 (c.962G>C.; p.Arg321Pro) (9). The second case was a childhood-onset LS in a Chinese male from a non-consanguineous family. The patient had had normal infancy and childhood, but his symptoms started at age 7 with progressive asymmetric dystonia starting in the right upper limb, followed by dysarthria and speech disturbance. No sign of intellectual disability was noted. Subsequent exome sequencing revealed a homozygous pathogenic missense variation in exon 11 of NDUFA9 (c.1078C>T c.1078C>T; p.Arg360Cys). At 28 years of age, Neuromuscular and Electrodiagnostic Testing (EDX) showed axonal motor and peripheral sensory polyneuropathy. Laboratory tests were remarkable for mild increases in postprandial lactate levels. Multiple MRIs revealed progressive bilateral T2 hyperintensities in the putamen and left caudate nucleus atrophy compatible with LS. His condition was stable in his mid-forties (11).
The phenotype of our patient was similar to the first case, with manifestations of lactic acidosis presenting in the first few weeks after birth. Moreover, both patients suffered from spells of epilepsy and apnea. While our patient had spasticity, the first reported case showed both spasticity and dystonia. The first case showed elevated alanine levels, whereas our patient showed a rise in BCAAs (leucine and isoleucine) and ammonia. The CT scan showed brain atrophy, hydrocephalus, and white matter hypodensity with no necrotic lesions in our patient (Figure 2); however, an MRI on the sixth day of birth in the first case revealed focal necrotic lesions. On the other hand, the second case had entirely different manifestations with a milder phenotype. Interestingly, his variation was very close to the site of variation in our patient, separated by only three amino acids; however, the severity of the clinical phenotype was significantly lower.

A spiral brain CT scan showed brain atrophy, hydrocephalus, and white matter hypodensity with no necrotic lesions
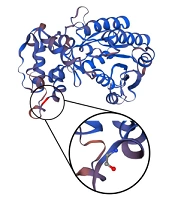
In the two previous cases, the pathogenicity of the variations was proven by functional studies (11) due to disruptions in the complex I assembly. This case report faces a particular limit due to the unavailability of functional studies in Iran. To address this issue, the heterozygous state of the identified variant was validated in the patient’s parents via Sanger sequencing (Figure 1D). Furthermore, we used variation predictor software (Table 1) to assess the pathogenicity of the patient’s variation: PolyPhen-2 (14) (http://genetics.bwh.harvard.edu/pph2) “probably damaging,” score: 1.000; VariationTaster (15) (http://www.variationtaster.org, build: NCBI 37/Ensembl 69): “disease-causing,” Amino acid alignment of the human NDUFA9 showed that the specific arginine involved in this variation is highly conserved through several species (Figure 1C). Constructing the three-dimensional view of the second structure of a protein using SWISS-MODEL Workspace/GMQE (16) showed that the changed amino acid is located between the end of a beta-helix and a turn (Figure 3). Moreover, since arginine (an essential amino acid) was changed to glycine (an aliphatic amino acid) in this variation, this change would likely lead to alterations in the structure of the entire protein.

Three-dimensional reconstruction of the second structure of the protein. The wild-type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions. The hydrophobicity of the wild-type and mutant residue differs, as well.
3.1. Conclusions
We reported a novel missense variant in NDUFA9, coding an accessory subunit of mitochondrial complex I, which caused a fatal Leigh-like syndrome in a four-month-old infant. Of two previously reported variations in NDUFA9 (9, 11), one was fatal, and the other led to patient survival into adulthood with stable disease. In these cases, the variations caused a defect in complex I assembly, with a more serious defect leading to a more severe clinical phenotype. Future studies are required to elucidate the association between the reported variations, protein function, and clinical phenotype.
