

1. Background
Glycine encephalopathy or nonketotic hyperglycinemia (NKH; Online Mendelian Inheritance in Man–OMIM no. 605899) is a rare, congenital, metabolism disorder with autosomal recessive transmission, which leads to an increase of glycine in all bodily fluids, especially in the central nervous system (CNS) (1). The metabolic defect is a lack of activity in the mitochondrial glycine cleaving enzyme complex (1-3). There are four forms of NKH, including neonatal, temporary, infantile, and late onset (4). Neonatal NKH emerges in the first few days of life with hypotonia, poor sucking, apnea, convulsions, and hiccups, and rapidly progresses to deep coma and generally death.
Despite aggressive supportive treatment, prognosis is generally poor. In surviving patients, severe neurodevelopmental retardation and uncontrollable convulsions are seen (4, 5). Together with an increase in the concentration of cerebral spinal fluid (CSF) glycine, CSF/serum glycine ratio > 0.08 supports the dianosis. Glycine cleavage enzyme activity or mutation studies can be made of liver or brain samples for a definitive diagnosis. Enzyme activity is close to zero in the neonatal type (2), which is estimated to have an incidence of approximately 1/250,000, although regionally high rates of 1/12,000 - 1/63,000 have been reported (6, 7).
2. Objectives
The aim of this study was to evaluate the clinical and laboratory findings and response to treatments in 10 patients diagnosed with neonatal NKH.
3. Methods
The study included patients diagnosed with neonatal NKH between August 2013 and July 2020. The sociodemographic characteristics, clinical and laboratory findings, treatments applied, and prognosis were evaluated retrospectively. The diagnoses of the patients were made from clinical findings and, at the same time, a CSF glycine/serum glycine ratio > 0.08, supported by neuroradiological imaging, electroencephalography (EEG) findings, or mutation analysis.
A checklist including age on presentation at hospital, gender, gestational age, birthweight, parental consanguinity, complaints, physical examination findings, clinical course, laboratory results (hematological and biochemical parameters, plasma amino acid and urine organic acid levels), plasma acylcarnitine profiles, tandem mass spectroscopy, CSF glycine/serum glycine ratio, EEG, brain magnetic resonance imaging/spectroscopy (MRI/S), mutation analyses, treatments used (sodium benzoate, phenobarbital, phenytoin, levetiracetam), and length of stay in hospital was prepared for each patient. Serum and CSF glycine levels were examined using the simultaneous high performance liquid chromatography (HPLC) method.
The study was approved by the Clinical Research Ethics Committee (decision no: 2020/278, dated: 23/09/2020).
4. Results
Out of 10 patients evaluated, five cases were female, and five were male with a median birthweight of 3,210 g (range: 2,750 - 4,000 g). One birth was premature, and nine were full-term. Parental consanguity was present in eight cases. In one case, there was a history of NKH in a sibling with mortality. The median age at admission was three days (range: 2 - 8 days), and the most common complaints were poor sucking, lethargy, and convulsions. In the physical examinations, evident hypotonia and reduced or absent neonatal reflexes were predominant and determined in all the patients. Convulsions were present in all the patients, and with the exception of two patients, had emerged in the first seven days.
The CSF/serum glycine ratio was > 0.08 in all patients, with median value of 0.19 (range: 0.12 - 0.30). The full blood count, C-reactive protein, kidney and liver function tests, urine organic analyses, and plasma acylcarnitine levels of the cases were within normal ranges. The EEG findings were normal in three cases, and a burst-suppression pattern was determined in seven cases.
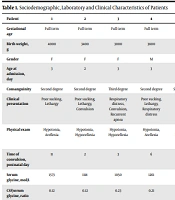
Brain MRI was taken in eight cases. The most common MRI findings were cortical atrophy and partial or complete agenesis in the corpus callosum. In five cases with MRS applied, two were normal, and there were glycine NAA and Cho peaks in three cases. For technical reasons, mutation analysis could only be performed in one case. Mechanical ventilation (MV) was required for nine patients because of respiratory failure and was applied for a median of 10 days (range: 2 - 28 days). Phenobarbitol was started as the first treatment option for convulsions. In patients where the convulsions could not be controlled, phenytoin was added, and less often levetiracetam. The resistant convulsions in one patient were controlled with dextromethorphan. As soon as diagnosis was made of probable NKH from the CSF/serum glycine ratio and supportive EEG, MR/MRS, and clinical findings, treatment was started using sodium benzoate (250 - 500 mg/kg/day) and a low protein diet (1 g/kg/day). With medical and supportive treatment, the enteral intake of seven patients reached a sufficient level. Patients who no longer required MV were discharged. Despite the medical and supportive treatment, mortality developed in three cases because of respiratory failure. Sociodemographic, laboratory, and clinical characteristics of patients are shown in Table 1.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Gestational age | Full term | Full term | Full term | Full term | Full term | Full term | Full term | Full term | Premature | Full term |
| Birth weight, g | 4000 | 3400 | 3000 | 3800 | 3700 | 3040 | 3140 | 2900 | 2750 | 3280 |
| Gender | F | F | F | M | M | F | M | M | M | F |
| Age at admission, day | 3 | 2 | 3 | 3 | 8 | 4 | 3 | 3 | 4 | 6 |
| Consanguinity | Second degree | Second degree | Third degree | Second degree | Second degree | Second degree | Unrelated | Unrelated | Second degree | Second degree |
| Clinical presentation | Poor sucking, lethargy | Poor sucking, lethargy, convulsion | Respiratory distress, convulsion, recurrent apnea | Poor sucking, lethargy, respiratory distress | Convulsion, hiccups | Poor sucking, lethargy respiratory distress, hiccups | Convulsion | Respiratory distress, recurrent apnea | Convulsion, hiccups | Poor sucking, lethargy, apnea |
| Physical exam | Hypotonia, areflexia | Hypotonia, hyporeflexia | Hypotonia, hyporeflexia | Hypotonia, areflexia | Hypotonia, hyporeflexia | Hypotonia, areflexia | Hypotonia, areflexia | Hypotonia, areflexia, low respiratory effort | Hypotonia, areflexia | Hypotonia, areflexia, low respiratory effort |
| Time of convulsion, postnatal day | 11 | 2 | 3 | 6 | 7 | 5 | 3 | 4 | 2 | 10 |
| Serum glycine, mol/L | 1573 | 1118 | 1050 | 1261 | 1130 | 1208 | 960 | 963 | 818 | 939 |
| CSF/serum glycine, ratio | 0.12 | 0.12 | 0.23 | 0.21 | 0.22 | 0.17 | 0.14 | 0.26 | 0.14 | 0.11 |
| EEG findings | Burst-suppression | Burst-suppression | N | Burst-suppression | Burst-suppression | N | Burst-suppression | N | Burst-suppression | Burst-suppression |
| Mechanical ventilation support, day | 10 | 4 | 20 | 18 | 11 | 5 | - | 28 | 2 | 2 |
| Age at discharge or death, day | 28 | 25 | 76 | 48 | 19 | 21 | 22 | 31 | 6 | 76 |
| Gene mutation | - | - | - | - | - | - | - | - | - | c. 1148 C > T homozigot |
| Outcome | Survive | Survive | Survive | Survive | Death | Survive | Survive | Death | Death | Survive |
Sociodemographic, Laboratory and Clinical Characteristics of Patients
5. Discussion
Neonatal NKH is the most common and severe form of glycine encephalopathy, with symptoms generally emerging in the first few days of life. Newborn infants characteristically present with hypotonia, lethargy, convulsions, and apnea, and are generally lost within the first year of life (5). In NKH, as one of the important examples of intoxication type congenital metabolism diseases, the majority of infants are normal at birth. Within a few days postnatally, hypotonia becomes evident, and with the loss of newborn reflexes, advanced encephalopathy develops (8).
With an inhibitor neurotransmitter-like effect on the brain stem and spinal cord, glycine can cause feeding difficulties, lethargy, hypotonia, apnea, and hiccups. Furthermore, with the excitator effect on N-methyl-D-aspartate (NMDA) receptors in the cerebral cortex, there may also be irritabillity and myoclonic seizures (9, 10). In a study that evaluated 65 cases, convulsions were seen in 75%; there was a need for MV support in two-thirds; 40% of the cases were lost in the neonatal period; and the survival rate and prognosis were reported to be better in males (11). Similarly, in the current study, the clinical status of infants with no problem at birth deteriorated in 2 - 8 days postnatally; hypotonia, lethargy, and progressive encephalopathy were seen; convulsions were observed in all the cases; and with the exception of one case, a need for MV was determined in nine other cases. In contrast to literature, all the exitus cases were male, which could be attributed to the limited number of patients evaluated.
In NKH, the increase in glycine in plasma, CSF, and urine is pathognomonic. Hyperglycinemia may also be determined together with ketoacidosis in organic acidemia (12). The level of CSF glycine in neonatal NKH may be as high as 30-fold more than normal (13, 14). The basic diagnostic criteria is a CSF/serum glycine ratio > 0.08 (normal < 0.02] together with the absence of organic acidemia (15). It is recommended that serum and CSF sampling is applied at the same time as far as possible. It must be taken into consideration that, especially in CSF samples, the presence of blood cells may change the result, and therefore care must be taken when obtaining the sample (3).
In all our cases, the serum and CSF glycine levels and the CSF/serum glycine ratio were much higher than normal values. Similar results were obtained in the control samples, which were taken to reduce technical or laboratory evaluation errors. No findings were determined in any of our patients suggesting organic acidemia (blood gas, urine organic acid, blood glucose, ketone, and acylcarnitine profile were all within normal limits).
Although a burst suppression pattern on EEG supports the NKH diagnosis, it is not accepted as a diagnostic method (16). Other than this pattern, hypsarrhythmia, multifocal epileptiform abnormalities, and sharp vertical waves may be seen on EEG (10). Burst suppression pattern was determined in most of our cases. Various MRI findings have been reported, including arachnoid cyst, intracranial bleeding, corpus callosum a/hypogenesis, delayed myelinisation in cerebral white matter, gyral malformations, ventricular expansion, hydrocephaly, and cerebellar hypoplasia (16). On MRS, no increase in myo-inositol together with glycine peak is a typical finding (17). In the current study, cortical atrophy was the most common finding, and in three of the five patients where MRS could be studied, glycine peak was determined. MRS can be useful in early diagnosis.
There is no current effective treatment option which can change the disease prognosis (10, 18). In literature, in contrast to studies that do not recommend diet treatment because glycine is a non-essential amino acid, there are other studies reporting that a limited protein diet can reduce glycine, and thus be used as a part of treatment; but no consensus has been reached on this subject (1, 10, 18).
The main aim of treatment is to reduce the high level of glycine in the CNS and inhibit the effects on neurotransmitters. The first option in treatment is sodium benzoate, which converts glycine to hippurate thereby providing excretion with the urine (5, 18). In 1986, Wolff et al. published the responses to treatment of high-dose sodium benzoate in three patients (one diagnosed at nine months old and the other two at birth) (19). Although seizures were controlled, positive effects were not observed on psychomotor retardation. This was reported to be due to the relatively late start of treatment or that the CNS level had not been lowered despite the increase in glycine excretion (18, 19). Dextromethorphan and ketamine, which are NMDA receptor antagonists, are other treatment modalities (9, 20).
Despite multiple anti-epileptic treatments, seizures in NKH may not be controlled. Successful results have recently been reported of the use of levetiracetam in resistant seizures in the neonatal period (10, 21, 22). This has important properties such as not making enzyme induction, not interacting with other anti-epileptics, and low binding to plasma proteins. İn addition, it does not lead to an increase in apoptosis in animal models unlike other anti-epileptics (23-25). In the current study, seizure not controlled efficiently in three cases with multiple anti-epileptic treatment were fully controlled with levetiracetam. For seizures resistant to treatment in NKH, the use of levetiracetam may be useful.
The main limitations of this study included: the retrospective nature of the study, a low sample size because NHK is a rarely seen metabolic disease, and genetic analysis was performed only on one patient.
In conclusion, for patients presenting in the neonatal period with a sudden, unexplained clinical deterioration, progressive encephalopathy, hypotonia, and seizures, especially when there is a history of parental consanguinity, NKH should be considered. Levetiracetam can be a treatment option in patients with resistant seizures.
