






1. Introduction
Our patient was a 6-month-old infant referred to our hospital with the chief complaints of fever, rapid breathing, and poor feeding from three days ago. There was no history of chocking, coryza, or gastrointestinal symptoms.
She was the first child of the family. The mother conceived this girl after six years of infertility by in-vitro fertilization (IVF). She was delivered through cesarean section at 37 weeks of gestation and was hospitalized for nine days due to respiratory distress. There was no specific complaint after discharge from the hospital until the age of five months. The mother mentioned poor weight gain and occasional coughs, which had gradually exacerbated in the last month. The weight upon referral was 5.2 kilograms, which was between 2nd and 5th percentile. Before admission to our hospital, she was referred to a pediatrician’s office, and sweat test was performed and repeated, which revealed borderline results. She had received Creon capsule, montelukast tablet, and fluticasone spray for one month because of suspected cystic fibrosis (CF). She was also receiving levothyroxine because of congenital hypothyroidism.
On physical examination, she was tachypneic (RR:70) and febrile (T:38). The oxygen saturation without supplemental oxygen was 85%. The lungs were clear on auscultation, and she had subcostal retraction. Other examinations were unremarkable.
She was admitted to the Emergency Department and received oxygen therapy by noninvasive ventilation (NIV), as a result of which oxygen saturation increased to 100%. The initial laboratory data are shown in Table 1.
| Variables | Data |
|---|---|
| Complete blood count | |
| WBC (cells per cubic millimeter) (Neutrophil / Lymphocyte) | 28000 (17/66) |
| Hemoglobin (g/dL) | 12.2 |
| Platelet (cells per microliter) | 482 |
| Biochemistry | |
| Sodium (mEq/L) | 130 |
| Potassium (mmol/L) | 3.3 |
| Blood glucose (mg/dL) | 89 |
| Blood urea nitrogen (mg/dL) | 13 |
| Creatinine (mg/dL) | 0.5 |
| Calcium (mg/dL) | 9.2 |
| Magnesium (mg/dL) | 2.2 |
| Inflammatory markers | |
| Erythrocyte sedimentation rate (mm/h) | 5 |
| C-reactive protein (mg/L) | 11 |
| Coagulative tests | |
| Prothrombin time (PT) (second) | 15.5 |
| International normalized ratio (INR) | 1.16 |
| Partial thromboplastin time (PTT) (second) | 32 |
| Arterial blood gas | |
| pH | 7.46 |
| PaO2 (mmHg) | 85 |
| PaCO2 (mmHg) | 50.8 |
| HCO3 (mEq/L) | 35.5 |
| Base excess | +10.4 |
Initial Laboratory Data
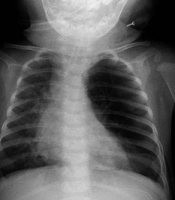
A chest X-ray (CXR) was performed, which revealed mild hyperlucency of the left upper lobe (LUL), with the rightward deviation of trachea and mediastinum (Figure 1).

First CXR, mild hyperlucency of the left upper lobe
Physiotherapy and antibiotic therapy were started. Treatment continued with the initial diagnosis of CF, but after one day, our patient’s respiratory condition deteriorated, and she was transferred to the Pediatric Intensive Care Unit (PICU) and underwent endotracheal intubation (ET). Another CXR was requested, which showed marked hyperlucency and overinflation of the LUL and resultant collapse of the left lower lobe along with mediastinal and tracheal shift to the right. The tip of the endotracheal tube was in the correct position, as reported by our radiologist (Figure 2). The radiologist also suggested further evaluation by spiral chest computed tomography scan (CT-scan) to rule out congenital lobar overinflation.

CXR after endotracheal intubation marked LUL hyperlucency
2. Arguments
2.1. Differential Diagnoses
Dr. Masoud. Mohammadpour (CMC pediatric intensivist): This infant was transferred to the PICU on the initial days of admission. I know the final diagnosis of this case; indeed, we had several challenges in managing her, and I want to share our experiences.
Dear Dr. Shirzadi, would you please help us with the differential diagnoses and approach to this patient?
Dr. Rohola. Shirzadi (CMC pediatric pulmonologist): The first CXR seems to be mildly rotated to the right side, as the distance of the medial head of clavicles to the vertebral spinous processes is not equal. In this situation, mild hyperaeration might be present on the left side. On the other hand, the heart size may not be measured reliably; in general, it is recommended to repeat the CXR for a more precise interpretation.
Based on these CXRs, the leading pathology might be on both sides. Cystic lesions, lung anomalies, airways obstruction, and mucus plaque (with check valve mechanism) can cause hyperaeration of the ipsilateral side and mediastinal shift to the contralateral side. Atelectasis and lung hypoplasia can deviate the mediastinum to the involved side and lead to compensatory hyperaeration in the other lung.
Furthermore, in the first ABG, bicarbonate level was 35.5 mEq/L, which shows the presence of a metabolic alkalosis component.
In this particular case, the following differential diagnoses could be raised.
2.1.1. CF Exacerbation and Mucus Plaque
Airway obstruction by mucous plaque frequently occurs in CF patients.
A thin layer of mucus in the healthy airway helps with a better lung function, but abundant airway secretion can cause bacterial overgrowth and airway obstruction. On the other hand, mucus plaque and subsequent neutrophilic inflammation can be present without the clinical presentation of obvious infection (1).
According to the history and suspicious sweat tests, which were taken after the treatment of hypothyroidism since untreated hypothyroidism can result in false-negative sweat test (2), CF exacerbation and subsequent airway mucus plaque were the most probable conditions explaining the patient’s symptoms, with atelectasis in the right lung and compensatory hyperinflation in the left.
2.1.2. Pneumothorax
Air retention in the pleural space causes pneumothorax, which can result in partial or complete lung collapse. In pneumothorax, hyperinflation occurs on the same side, and in cases of tension pneumothorax, mediastinal shifts may occur on the opposite side. In this patient, due to the fact that hyperinflation occurred on one side and the mediastinal shift on the opposite side, pneumothorax can be a differential diagnosis.
2.1.3. Foreign Body Aspiration
Foreign body aspiration is one of the most life-threatening pediatric respiratory emergencies, since it can lead to complete airway obstruction. Aspiration of a foreign body may occur with no obvious history or without any evidence at the time of the aspiration, in which case early diagnosis needs high index of suspicion. Chest X-ray is typically recommended first; however, bronchoscopy should be carried out promptly for foreign body removal in the presence of positive history.
Due to the inability to walk in this young infant and lack of a precise history of aspiration, the possibility of foreign body aspiration was not high.
2.1.4. Congenital Lobar Emphysema
Congenital large hyperlucent lobe (CLHL) or congenital lobar emphysema is an infrequent lung disease characterized by air inflation in the involved lobe(s), the resultant compression of the proximate lung tissue, and mediastinal shift to the opposite side. It typically presents with respiratory distress in early infancy (3). In an infant with respiratory distress, unilateral lung hyperinflation, contralateral atelectasis, and mediastinal shift, CLE can be suggested; however, in older patients, it is necessary to rule out other differential diagnoses. Computed tomography scan and MRI can help in diagnosis. In this particular patient, CLE can be suggested due to the early onset of cough and respiratory distress and consistent radiologic findings.
2.1.5. Vascular Ring
Congenital abnormalities of the aorta and its main branches can result in vascular ring formation and compression around the trachea and esophagus. Respiratory and gastrointestinal symptoms may then follow, such as stridor, wheezing, swallowing disfunction, and chocking (4). As a result of a partial bronchi encirclement, check valve mechanism may occur with subsequent air trapping on the same side and atelectasis and mediastinal shift to the opposite side.
2.1.6. Extrinsic or Intrinsic Airways Compression
In the presence of mediastinal mass, bronchogenic cyst, granulation tissue, or any other intrinsic or extrinsic compression, ipsilateral hyperinflation may occur with the same mechanism as vascular rings.
2.1.7. Swyer-James or Macleod Syndrome
In this disorder, an infectious insult causes decreased lung vascularity and lung hyperaeration (5). As this condition is usually found in older patients, it could be ruled out in our patient.
2.1.8. Poland Syndrome
This rare congenital syndrome presents with the unilateral absence of pectoralis muscles along with other abnormalities of the breast, nipple, and upper extremity. Radiologic findings include unilateral hyperlucent area on the affected side (6). As this patient had normal appearance without any chest wall asymmetry, it could be ruled out.
2.1.9. Mounier-Kuhn Syndrome and Williams-Campbell Syndrome
Mounier-Kuhn syndrome is characterized by marked dilation in tracheobronchial airways because of the thinning of muscles. In Williams-Campbell syndrome, there is cartilage defect of the airways.
In these conditions, bronchiectasis develops and presents with frequent respiratory infections and recurrent cough. Computed tomography scan can be helpful in diagnosis (7, 8).
To approach such cases, precise history taking and physical examination are needed. Chest X-ray is typically the first-line imaging and sometimes sufficient for diagnosis; however, it is better to confirm the diagnosis with further imaging. Computed tomography scan might show the potential extrinsic causes of obstruction, vascular anomalies (in contrast imaging), and emphysematous segments and lobes and its severity. Sometimes macrocystic forms of congenital cystic adenomatoid malformation (CCAM) are presented as an emphysematous lobe on CXR, and CT scan is helpful in differentiation. The differentiation is critical as conservative management has no place for CCAM.
Bronchoscopy might be helpful to rule out extrinsic airway compression, vascular rings, foreign bodies, mucus plug, meconium mucus plug (in newborns), granuloma, cartilage abnormalities, and bronchus atresia; however, it is not necessary in all cases.
Ventilation / perfusion scan might also be helpful, but negative results do not exclude the abnormality of perfusion and ventilation.
Echocardiography is also suggested to evaluate the possibility of associated anomalies and vascular causes of bronchial tree obstruction.
2.2. Further Evaluation
After pulmonary consultation, bronchoscopy was performed, which showed only abundant and thick secretions, especially in the left main bronchus, which were removed. Cardiology consult was done and revealed normal cardiac function and no major structural abnormalities. Due to the current COVID-19 pandemic, COVID-19 PCR was requested and was negative. The patient was a candidate for a contrast CT scan, but the respiratory condition deteriorated despite high setup mechanical ventilation. An ABG was then immediately obtained, which revealed severe respiratory acidosis.
pH:6.98, PCO2:109 mmHg, HCO3:24.7 mEq/L, BE:-5.9
After a team consult, the patient was a candidate for emergency thoracotomy. The surgeon reported that the LUL was extremely emphysematous, and the left lower lobe was collapsed. Thus, CLE was grossly confirmed, and partial lobectomy was performed (Figure 3).

The markedly emphysematous lobe and readily visualized individual alveoli
She was returned to the PICU while she was intubated and had bilateral chest tubes. In the first CXR, which was done a few hours after thoracotomy, bilateral pneumothorax subsequent to surgery was seen. After a few days, the right chest tube was removed, then the patient was weaned from mechanical ventilation, and NIV was used. Her respiratory condition improved gradually. The other chest tube was removed a few days later, and she was weaned from supplemental oxygen. She tolerated the condition with no respiratory distress and maintained normal oxygen saturation at room air. She was then transferred to the surgery ward. Because of the complicated post-operative period and her previous medical history, sweat test was requested. Sweat sodium and chloride were 70 and 95, respectively. Molecular genetic for the CFTR gene was requested, and no mutation was detected. Lung pathology confirmed emphysema (Figure 4).

The microscopic examination of the hematoxylin and eosin-stained sections shows a uniform overdistension of apparently normally developed acini. The focal disruption of alveolar walls is noted. The examination of the bronchus of the lobe reveals no stenosis, atresia, or intrinsic obstruction. Cartilage abnormalities of the bronchial wall, Hyaline membrane, or thick secretions are not appreciated.
Finally, she was discharged from our hospital on the 21th post-operative day, with recommendation to close follow-up.
2.3. Clinical Diagnose
Congenital lobar emphysema.
2.4. Discussion
2.4.1. Discussion of Management
Congenital lobar emphysema is one of the congenital cystic lung lesions that leads to the overdistension of one or more pulmonary lobes (9). Its prevalence ranges from 1 in 20000 to 1 in 30000. It mostly affects the left upper lobe and then the right middle, right upper, and lower lobes, respectively (10). About half of the cases present in the neonatal period by respiratory distress and the other in the first six months of life (11, 12); presentation after infancy is rare. The condition is sometimes associated with cardiac anomalies; thus, echocardiography is reasonable, as we did in our patient (13).
Karnak et al. conducted a study in Turkey in 1999 and designed an algorithm for the approach to CLE (14). They suggested CXR as the first-line imaging and recommended performing CT-scan, ventilation-perfusion scan, and bronchoscopy in a combination, depending on the clinical scenario of the patient to confirm the diagnosis.
On the other hand, for children under two months of age, surgical lobectomy is suggested as soon as possible, and conservative management has no place. Similarly, the treatment of choice for CLE in patients younger than two months of age or any child with severe symptoms is lobectomy, and conservative management is reserved for older children with mild symptoms and close follow-up for the progression of symptoms and possible need for surgery (15).
In literature, bronchoscopy in patients with CLE and severe respiratory distress is controversial and might be harmful. It is reserved for older infants who are candidate for conservative management and for the exclusion of foreign body (12). Thus, based on the age and medical history of the patient (suspicious of CF), bronchoscopy was indicated; however, the respiratory condition of the patient deteriorated after the procedure.
Dr. Hojatollah Raji (CMC Pediatric Surgeon) commented:
If the child is asymptomatic and has a supportive family, he/she could be managed conservatively with close follow-up. In symptomatic patients, we should resect the emphysematous lobe, and no other treatment is reasonable. However, before surgery, CT scan is highly recommended because it reveals more precise information and is more specific. In cases of suspected foreign body aspiration or obstruction by respiratory secretions and mucus plugs, bronchoscopy might be helpful.
If the patient is stable, respiratory distress is mild to moderate, and oxygen saturation and ABGs are acceptable, we should absolutely do further imaging before surgery; however, in the presence of severe respiratory distress and life-threatening respiratory acidosis, as was the case with our patient, we cannot waste time for further diagnostic evaluations. Accordingly, we were obligated to do the surgery to save the patient’s life, which was one of the strength of the approach to the patient. In cases of CLE, the emphysematous lobe suddenly protrudes from the surgical field as a balloon and the diagnosis of CLE is grossly confirmed (Figure 3).
2.4.2. Discussion of Cystic Fibrosis
Dr. Rohola. Shirzadi (CMC pediatric pulmonologist): The gold standard for the diagnosis of CF is sweat test (16). This patient underwent sweat test three times; two tests had borderline results, and the last one was positive (chloride = 95 mEq/liter). On the other hand, malnutrition and FTT result in false-positive sweat test.
Based on Nelson’s textbook of pediatrics (17), two positive sweat tests are necessary for the definite diagnosis of CF.
In CF, the lungs are destructed over time, and bronchiectasis develops gradually, so resecting a part of the lungs may be extremely harmful, and we should do our best to reserve the lungs as much as possible. This patient had life-threatening respiratory acidosis, and lobectomy was the only lifesaving option. Decision-making in such complicated situations is really challenging. If the patient’s condition was slightly better, giving another chance to patient might have been reasonable before lobectomy.
Untreated hypothyroidism can cause false-negative sweat test results (2); that is why it is better to recheck the patient’s thyroid hormones status.
2.4.3. Pathologic Discussion
Dr. Aileen Azari-yam (CMC Pathologist): The main pathological differential diagnosis includes cystic adenomatoid malformation type III, which has similar findings on ultrasound. Congenital lobar emphysema is devoid of cystic lesions as compared to cystic adenomatoid malformation (18, 19).
Currently, more than 2000 disease-causing CFTR mutations have been reported in databases, which vary in their frequency and distribution in different populations. In the European population, about half of these mutations are rare variants. A very small number of mutations have a global frequency more than 0.1%; however, some of them can account for the high percentage of mutations in certain populations (20).
Our patient was screened by CF StripAssay (Viennalab diagnostics, GmbH, Austria), which detects 34 common Caucasian CFTR mutations and the IVS8 variants 5T/7T/9T. No mutation was found by this assay. The detection rate for this test was estimated to be 38% in a study from Isfahan Province (21). More comprehensive studies are needed addressing the mutation detection rate of this test in the Iranian population.
The gold standard method for the diagnosis of CFTR mutations has been Sanger sequencing (22), which was suggested for our patient.
Mutation detection and variant analysis could be quite complex in cystic fibrosis. The combined use of all scanning / screening and direct techniques (eg, DGGE, DHPLC, HRMCA, ARMS-PCR, Sanger sequencing, etc.) cannot give assurance to find both mutated alleles in every CF patient, and 1 - 5% of alleles are not identified. Lots of CFTR mutation tests cannot detect CFTR regulatory mutations far from the genes or located in non-coding regions of the gene (23).
Multiple next-generation sequencing (NGS)-based targeted panels are available for the mutation detection of the CFTR gene. Illumina next-generation sequencing (NGS) products for CF evaluation are approved by the FDA. The TruSight Cystic Fibrosis 139-Variant Assay finds as many as 139 CFTR variants in the CFTR2 database and supports the best clinically and functionally applicable variants in a disparate community (24, 25).
3. Conclusions
In conclusion, we should consider CLE as one of the life-threatening causes of severe respiratory distress in infants presenting with local hyperaeration on CXR. In case of severe respiratory symptoms, we suggest surgical treatment without further imaging. In mild to moderate respiratory symptoms, conservative or surgical management could be considered.
3.1. Take-home Messages
(1) The diagnosis of CLE can be made based on its characteristic appearance on CXR.
(2) Symptomatic patients in the first two months of age should undergo surgical treatment.
(3) Surgical management is indicated in any CLE patient with severe symptoms.
3.2. Follow-up
The patient was referred to the surgery clinic one week after discharge. She was stable and uncomplicated. We asked the parents to do whole exome sequencing for the definite diagnosis of cystic fibrosis, but they refused to do further evaluations.
3.3. Final Diagosis
According to the pathology report, congenital lobar emphysema was the definitive diagnosis, but we still have to rule out cystic fibrosis as a probable concomitant disease.
