

1. Background
Acute leukemia is one of the most common malignant tumors in children, occupying the first place in childhood cancer mortality. Of these, acute myeloid leukemia (AML) accounts for 20% to 30%. At present, the clinical treatment of AML still mainly relies on combination of chemotherapy and hematopoietic stem cell transplantation (HSCT), but the adverse reactions of chemotherapy are extremely grave. Therefore, exploring new ways to treat leukemia has become an urgent task for the effective treatment of leukemia.
Iron is one of the basic nutrients required for cell viability. Deferoxamine (DFO) is a widely used iron chelator in the clinic, and it is often used in the treatment of siderosis. In recent years, it has been reported that DFO can inhibit tumor cell proliferation and induce apoptosis (1-4), suggesting that DFO may be used as an anti-tumor agent. Indeed, DFO can inhibit the proliferation of prostate cancer, breast cancer, neuroblastoma, and leukemia cells (5, 6). Callens et al. proved that iron metabolism was an effective target for the treatment of AML (7). According to a study, DFO significantly enhanced the activity of dexamethasone, doxorubicin, and L-asparaginase, thus mitigating the multidrug resistance of leukemia (8). DFO also induced an increase in intracellular calcium, enhancing the sensitivity to doxorubicin as a chemotherapeutic drug (9).
Mitochondrial autophagy has become a research hotspot in recent years. Lemasters found that the decrease in mitochondrial membrane potential and the opening of mitochondrial permeability membrane pores could cause mitochondrial autophagy, and he proposed the concept of mitochondrial autophagy (10). Studies have shown that Parkin and Pink1 proteins are involved in the execution of mitochondrial autophagy induced by a decrease in membrane potential (11, 12). Therefore, the specific mechanism of mitochondrial autophagy can be elucidated by detecting the expression of these proteins.
2. Objectives
At present, there are few studies on mitochondrial autophagy in leukemia cells in the literature. In an in vivo experiment, the effect of DFO on the growth of HL-60 xenograft tumors was investigated in nude mice. On the eleventh day after inoculation, the inhibition rate of transplanted tumors was 2.67% in the DFO group (13). Therefore, we intend to prove that DFO could induce apoptosis and mitochondrial autophagy in HL-60 cells.
3. Methods
3.1. Materials
The HL-60 cell line was purchased from Beina Chuanglian Biotechnology Research Institute (Beijing, China). DFO was purchased from Sigma Company. RPMI-1640 medium and fetal bovine serum were purchased from Gibco Company. Cell counting Kit-8 (CCK-8) was purchased from Dongren Chemical Technology Company (Shanghai, China). Hoechst 33342 was purchased from Yuheng Biotechnology Company (Suzhou, China). An annexin V-APC/7-AAD double-staining apoptosis detection kit was purchased from Kaiji Biotechnology Development Company (Nanjing, China). The reactive oxygen species (ROS) detection kit was purchased from Solarbio Biotechnology Company (Beijing, China). The rabbit primary antibodies against heat shock protein 60 (HSP60), Nip-like protein X (NIX), and microtubule-associated protein 1 light chain 3 (LC3) used for western blotting were purchased from Cell Signaling Technology (Massachusetts, USA). Antibodies against translocase of inner mitochondrial membrane 23 (TIMM23) and translocase of the outer mitochondrial membrane member 20 (TOMM20) were purchased from Abgent Biotechnology Company (California, USA), and the sheep anti-rabbit secondary antibody was purchased from Proteintech Company (Wuhan, China). Radioimmunoprecipitation assay (RIPA) lysis buffer, eECL Western Blot kit, BCA protein assay kit, HiFiScript cDNA synthesis kit, and UltraSYBR mixture were purchased from Kangwei Century Biotechnology Company (Wuhan, China). RNAiso Plus was purchased from Takara Company (Wuhan, China).
3.2. Cell Culture
HL-60 cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum and 1% penicillin-streptomycin in a carbon dioxide incubator at 37°C. The cells were collected for experiments in the logarithmic growth phase. The experimental groups were cultured in the presence of DFO at final concentrations of 25, 50, 100, and 200 μmol/L, while the control groups were treated with phosphate buffer solution (PBS).
3.3. Cell Viability Detection
Cell viability was assayed using Cell Counting Kit-8 according to the manufacturer’s instructions. Briefly, cells were seeded into 96-well plates and treated with DFO at concentrations of 10, 50, 100, and 200 μmol/L. The control groups received the same amount of PBS. After a pre-determined period of incubation, CCK-8 solution was added to the cells and incubated for an additional 4 hours. The optical densities of the plates were read with a microplate reader at 450 nm. Controls were set to determine the background absorbance.
3.4. Hoechst 33342 Staining
HL-60 cells were seeded into 6-well plates (2 × 105/well) and treated with DFO at concentrations of 25, 50, 100, and 200 μmol/L for 24 hours in a cell incubator. The control groups received the same amount of PBS. Nucleic acid staining was performed with Hoechst 33342 according to the manufacturer’s instructions. Briefly, 1 mL of staining solution was applied to each well of 6-well plates to cover the samples completely; they were stained in the dark for 20 minutes. The stained samples were observed under a fluorescence microscope.
3.5. Apoptosis Analysis
In the pre-test, we found that the drug had the greatest impact at 200 μmol/L; thus, we chose 200 μmol/L for the experiment. HL-60 cells were seeded into 6-well plates (2 × 105/well) and cultured with DFO at a concentration of 200 μmol/L for 24 hours in a cell incubator. The control groups were treated with the same amount of PBS. Apoptosis was detected using an annexin V-APC/7-AAD apoptosis detection kit according to the manufacturer’s instructions. Briefly, cells were collected and resuspended in 500 μL of binding buffer. Then, the cells were incubated with 5 μL of annexin V-APC and 5 μL of 7-AAD for 15 minutes in the dark. The apoptosis was analyzed by flow cytometry.
3.6. Detection of ROS Level
HL-60 cells were treated with DFO at concentrations of 25, 50, 100, 200, and 400 μmol/L for 24 hours. The control groups received the same amount of PBS. The 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) probe was loaded according to the manufacturer’s instructions. Then, the cells were incubated for 30 minutes at 37°C and with inversion every 5 minutes; thus, the probe was in full contact with the cells. Subsequently, the cells were washed in serum-free RPMI-1640 medium 3 times to completely remove the DCFH-DA that did not enter the cells. The fluorescence of the cells was detected by flow cytometry.
3.7. Western Blot
The cells were inoculated in 6-well plates (5 × 106) and cultured for 24 hours. Then, DFO at concentrations of 25, 50, 100, and 200 μmol/L was added to plates, and cells were incubated for the indicated amount of time (6, 12, 24, and 48 hours). The control groups received the same amount of PBS. Total protein was extracted from HL-60 cells by RIPA lysis buffer supplemented with a 1% phosphatase inhibitor cocktail on ice. After centrifugation at 14 000 g for 10 minutes at 4°C, the supernatant was collected, and the protein was quantitated by a BCA protein assay kit. Equal amounts of protein were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with 5% nonfat milk, the membranes were washed 3 times with tris-buffered saline and tween 20 (TBST) and incubated overnight with primary antibodies. Then, the membranes were rinsed in TBST and incubated with corresponding secondary antibodies for 2 hours. The bands were visualized with an eECL western blot kit. The membranes were placed in a chemiluminescence imager to observe and take pictures.
3.8. Quantitative Real-Time Polymerase Chain Reaction
The cells were inoculated in 6-well plates (5 × 106) and cultured in an incubator for 24 hours. Then, DFO at a concentration of 200 μmol/L was added to the culture for 24 hours. The control groups received the same amount of PBS. Total RNA was extracted from cells using RNAiso Plus according to the manufacturer’s protocol. RNA was reverse transcribed into complementary DNA (cDNA) with a HiFiScript cDNA synthesis kit. The messenger RNA (mRNA) levels were measured on a quantitative real-time polymerase chain reaction (PCR) instrument using the UltraSYBR mixture. The primers were designed by Jin Wei Zhi Biotechnology Company (Guangzhou, China). The relative mRNA level was calculated using the 2-ΔΔCt method.
3.9. Statistical Analysis
Each experiment was repeated 3 times, and the results are presented as the mean ± SD. Differences between the 2 groups were analyzed using the student t-test. Significance was defined as P < 0.05, *P < 0.05, and **P < 0.01.
4. Results
4.1. DFO Induced Apoptosis in HL-60 Cell Line
4.1.1. Cell Viability Detection
The cell viability was measured by CCK-8. The cell survival rate was calculated according to the formula and plotted. The effect of DFO on HL-60 was time-concentration dependent. In Figure 1, the survival rate of HL-60 cells decreased significantly after treatment with DFO, indicating that DFO inhibited the growth of HL-60 cells.

Detection of cell viability
4.1.2. Hoechst 33342 Staining
After adding Hoechst 33342 working solution and incubating for 20 minutes in the dark at room temperature, the cells were observed and photographed under an inverted fluorescence microscope. In Figure 2, the fluorescence intensity of apoptotic cells became stronger with the increase of DFO concentration compared with the control, and the nuclei were stained densely in fragments in Figure 2F. The results showed that apoptosis became obvious with the increase in DFO concentration.

Hoechst 33342 staining. A, The white light of the control group; B, Fluorescence of the control group; C, Fluorescence of HL-60 treated with deferoxamine at 25 μmol/L; D, Fluorescence of HL-60 treated with deferoxamine at 50 μmol/L; E, Fluorescence of HL-60 treated with deferoxamine at 100 μmol/L; F, Fluorescence of HL-60 treated with deferoxamine at 200 μmol/L.
4.1.3. Apoptosis Analysis
The cells were stained according to the instructions of the annexin V-APC/7-AAD double staining kit and detected by flow cytometry. In Figure 3, the apoptosis rate of cells was 15.6% in the control group, which was the sum of the fraction of cells in quadrants Q2 and Q4, while it was 18.4% in the experimental group. Compared with the control group, the fraction of apoptotic cells in the experimental group was higher, and we concluded that DFO promoted apoptosis.

Apoptosis detection by annexin V-APC/7-AAD double staining
4.2. DFO Induced Mitochondrial Autophagy in the HL-60 Cell Line
4.2.1. Detection of ROS Level
The DCFH-DA probe was loaded according to the instructions of the ROS detection kit. After incubating for 30 minutes at 37°C, cell fluorescence was immediately detected by flow cytometry, and fluorescence intensity data were obtained. Compared with the control group, the expression of ROS increased gradually with increasing DFO concentration, as shown in Figures 4 and 5, reflecting the increase in mitochondrial autophagy.

Expression of reactive oxygen species in HL-60 cells treated with deferoxamine at different concentrations.

Effects of deferoxamine at different concentrations on the production of reactive oxygen species in HL-60 cells.
4.2.2. Western Blot Detection
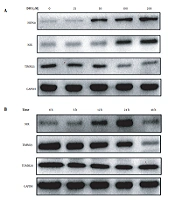
The expression of the mitochondrial autophagy proteins NIX and HSP60 increased, while the expression of TIMM23 decreased with increasing DFO concentration (Figure 6A). Additionally, when HL-60 cells were treated with 200 μmol/L DFO from 6 to 48 hours, the expression of NIX protein increased gradually and reached a peak at 24 hours, while the expression of TIMM23 protein decreased gradually and the expression the of TOMM20 protein did not show any significant change (Figure 6B).

A, Effects of deferoxamine at different concentrations on the expression of mitochondria-related proteins; B, Effects of deferoxamine at different time points on the expression of mitochondria-related proteins.
4.2.3. Detection of Quantitative Real-Time PCR
With GAPDH as the internal reference gene, fluorescence quantitative PCR detection showed that the expression of NIX mRNA and LC3 mRNA in HL-60 cells increased significantly, while the expression of TIMM23 mRNA decreased in the experimental group compared to the control group (Figure 7).

A, Expression of Nip-like protein X messenger RNA in HL-60 cells induced by deferoxamine at 200 μmol/L; B, Expression of microtubule-associated protein 1 light chain 3 messenger RNA in HL-60 cells induced by deferoxamine at 200 μmol/L; C, Expression of the translocase of inner mitochondrial membrane 23 messenger RNA in HL-60 cells induced by deferoxamine at 200 μmol/L.
5. Discussion
The etiology of leukemia has not been completely clarified thus far, but it may be related to physical, chemical, and genetic factors. For more effective treatment, targeted therapy can selectively kill tumor cells, which is a promising development (14, 15). Therefore, there is an urgent need to find new markers for early diagnosis, as well as to use them as targets for treatment to improve the overall prognosis of patients.
Mitochondrial autophagy has been an active research topic in recent years, and the role of mitochondrial autophagy in hematological tumors has received increasing attention. In the current project, we detected changes in the expression of mitochondria-related proteins, reflecting the activity of mitochondrial autophagy. We selected the mitochondrial autophagy-related pathway protein NIX, mitochondrial matrix protein HSP60, mitochondrial outer membrane protein TOMM20, and mitochondrial inner membrane protein TIMM23 to examine the changes in mitochondrial autophagy. Through a series of tests, we proved that DFO induced apoptosis and mitochondrial autophagy in HL-60 cells. Our team has previously found that after HL-60 cells were treated with DFO for 12, 24, 48, and 72 hours, the mitochondrial membrane potential (ψ m) decreased and that the degree of decrease was dose- and time-dependent. It was suggested that the apoptosis pathway mediated by mitochondria was the possible mechanism of iron deprivation-induced apoptosis in HL-60 cells. Combined with the latest research, we detected the evidence of mitochondrial autophagy, further proving that DFO induced apoptosis of HL-60 cells through mitochondrial autophagy. The apoptosis pathway mediated by mitochondrial autophagy was the possible mechanism of HL-60 cell apoptosis induced by DFO. This provides another experimental basis for the clinical use of DFO in the treatment of leukemia.
In the current project, we found that the levels of the mitochondrial autophagy-related pathway protein NIX as detected by western blotting changed in a concentration- and time-dependent manner upon treatment with DFO. The expression of NIX mRNA detected by real-time fluorescence quantitative PCR was consistent with the western blot results. NIX is a member of the Bcl-2 family and has the highest homology with BNIP3. NIX contains the apoptosis effect domain and transmembrane domain and can interact with Bcl-2 and Bcl-XL, which can change the permeability of the mitochondrial membrane, increase the release of cytochrome C, and cause apoptosis. NIX is closely related to mitochondrial autophagy. The NIX protein may be a new target for the treatment of leukemia.
There have been many studies on the mechanism of mitochondrial autophagy. However, in our previous literature search, we found that there were few studies on mitochondrial autophagy in leukemia, which led to some defects in our research. Western blot and quantitative real-time PCR detected only part of the protein. The effect of other protein drugs was not significant, and even some proteins were not detected. However, through experiments, we could still prove that DFO can induce apoptosis and autophagy and further induce apoptosis by inducing autophagy in leukemic cells. Various molecules are involved in the mechanism of mitochondrial autophagy. An in-depth study of the regulatory mechanism of mitochondrial autophagy helps understand the molecular mechanism of many diseases and lays a theoretical foundation for the development of targeted drugs.
