




1. Background
Limb-girdle muscular dystrophy (LGMD) is a genetic disease associated with heterogeneous clinical muscular symptoms such as shoulder and pelvic girdle weakness, characterized by progressive muscle weakness, proximal muscle atrophy, elevated creatine phosphokinase, and dystrophic findings in muscles. The disease prevalence varies greatly based on geography. The disease can be inherited as autosomal recessive (AR), autosomal dominant, or x chromosome. The disease incidence at younger ages is associated with a more severe phenotype of the disease (1, 2). A study in 2016 showed that the prevalence of LGMD was 1.6 per 100,000 (3).
Mutations in proteins responsible for cellular consistency, such as extracellular matrix, nuclear membrane proteins, and sarcomere proteins, are the main causes of complications in this disease, and collagen structure disorder is one of the other damages (4, 5). Various mutations in this gene associated with dystrophy in different populations show the extent of clinical manifestations of this disease (6). Various subtypes of this disease have been identified so far (7, 8). LGMD2A was the first LGMD in which a molecular-level disorder was shown (9). An astounding variety of genetic causes has been recorded in LGMD, so the identification of its types has gone beyond the traditional classification system (8). The prevalence of the disease varies greatly based on the geography of the residence place. In Denmark, unlike the rest of the world, LGMD2A is rare, and LGMD2I is prevalent (9-13).
It is difficult to differentiate between various forms of the disease, as its initial manifestations and muscle biopsy can be similar. However, some clinical information such as creatine kinase (CK) level, pattern of muscle involvement clinically and on MRI, and onset time of cardiac and respiratory involvement is helpful in differentiating subgroups (14, 15). Clinical manifestations vary based on the mutated gene. Clinical manifestations in alpha sarcoglycanopathies usually begin in the first decade of life, progress rapidly, and lead to wheelchair dependence within a few years. Leg hypertrophy is one of the symptoms of sarcoglycanopathies. Unlike sarcoglycanopathies, calpainopathies usually begin at the ages of 8 - 15, progress more slowly, and lead to wheelchair dependence 15 - 20 years after the onset of the disease. Leg hypertrophy is rarely seen in these patients, and if present, it is milder than alpha sarcoglycanopathies and Duchenne muscular dystrophy (16). Determining the clinical manifestations of various forms of the disease can help with differential diagnoses in each subgroup (17).
Reports show that few studies have been done on genetic characteristics, and knowing more about the disease from this point of view can help with LGMD treatments (18, 19). In addition, investigating genetic characteristics and their relationship with the severity of this disease can help uncover the underlying cause of this problem and identify more effective molecular and gene therapies in the future (20, 21).
2. Objectives
This study aimed to determine the genetic diversity and relative frequency of various forms of LGMD in Iranian children.
3. Methods
3.1. Study Design
In this descriptive research, 72 children suspected of inherited muscular diseases in the neurology or emergency department of the Pediatric Medical Center were studied from April 2019 to April 2020. All eligible children under 14 years of age were included in the study according to the inclusion and exclusion criteria. After preparing the informed consent form, parents' consent for participating in the research was obtained.
3.2. Inclusion and Exclusion Criteria
Inclusion criteria were at least 2 of the following: (a) clinical phenotype consistent with the diagnosis of LGMD (onset in the pelvic or shoulder girdle or both at the same time and early symptoms usually limited to one of these muscles); (b) findings of muscle biopsy consistent with myopathy or dystrophy; (c) the presence of mutation causing the disease in one of the LGMD genes; and (d) signing the informed consent document before participating in the study.
Exclusion criteria were different clinical manifestations from LGMD (distal onset of the disease and involvement of the face or extraocular muscles at the onset of the disease) or genetic or histopathological findings suggesting other neuromuscular diseases. Also, patients whose diagnosis was not confirmed by muscular or genetic biopsy were excluded from the study.
3.3. Description and Collection of Information
Patients' characteristics had been registered in the bioregistry system of neuromuscular diseases. Additional tests (muscle biopsy and genetic test) were performed to confirm the diagnosis. After confirmation of the diagnosis, the patients were examined for variables such as age, gender, type and level of disease, duration, creatine phosphokinase, contracture, cardiac involvement, respiratory involvement, history of kinship, and mutation in the genes responsible for the disease.
Information was collected using a checklist, including information on age, gender, symptoms and duration of disease, creatine phosphokinase (CPK) level, contracture, heart involvement, respiratory involvement, muscle biopsy, and muscles involved. A pathologist prepared the checklist. In order to confirm the data, the experiments were repeated once more.
3.4. Analysis of Gene Mutations
Specimens of muscle biopsies were analyzed by multiple Western blots using a mixture of monoclonal antibodies against α‐sarcoglycan (diluted 1:300), dystrophin (Dys‐2, diluted 1:1000), calpain‐3 (Calp12A2, diluted 1:800), and dysferlin (Hamlet, diluted 1:1000) (all purchased from Novocastra, Newcastle Upon‐Tyne, UK). The protein quantity of each sample was normalized to the amount of tissue loaded, which was determined by the presence of skeletal myosin bands in the Coomassie blue-stained gels after transfer. Muscle biopsies from patients showing normal protein levels by conventional immunoblotting underwent a further biochemical assay to test calpain‐3 autocatalytic activity as described previously (22).
3.5. Data Analysis Method
The data are presented using descriptive statistics, including frequency and mean (standard deviation), and drawing graphs and tables. Independent t-test and chi-square test were used to analyze the data (assessing the relationship between type of mutation and CPK level). SPSS version 22 software was used in all analyses.
4. Results
Out of the 60 patients suspected of limb-girdle muscular dystrophy, 19 were clinically suspected of LGMD and were excluded from the study (10 patients had nonspecific muscle biopsies, one patient had nonspecific genetic tests, and eight patients did not have muscle biopsies or genetic tests). Forty-one patients with a mean age of 11.11 were studied (the minimum age was 3 years and 11 months, and the maximum age was 36 years). Twenty-two (36.7%) patients were confirmed by a genetic test, and 19 patients were confirmed by muscle biopsy (Table 1).
| Parameters | Positive Genetic Test | Positive Biopsy | Total |
|---|---|---|---|
| Mean age (y) | 5.6 | 13.5 | 11.11 |
| Gender | |||
| Male | 9 (47.4%) | 13 (59.1%) | 22 (53.7%) |
| Female | 10 (52.6%) | 9 (40.9%) | 19 (46.3%) |
| Total | 19 (100%) | 22 (100%) | 41 (100%) |
Distribution of Age and Gender According to the Diagnostic Method in the Studied Patients
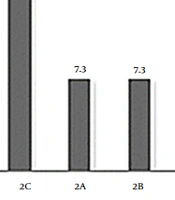
Concerning limb-girdle types, 11 (26.8%) patients had 2D (alpha sarcoglycanopathy), 10 (24.4%) patients had 2E (beta sarcoglycanopathy), 7 (17.1%) patients had 2C (gamma sarcoglycanopathy), 3 (7.3%) patients had 2A (calpainopathy), 3 (7.3%) patients had 2B (dysferlinopathy), 3 (7.3%) patients had dystroglycanopathy, 3 patients had titinopathy (2J), and 1 patient had laminopathy (Figure 1).

Frequency of limb-girdle types
Also, 68.3% had parental kinship. There was a history of similar disease in the family of 39% of patients. The initial symptoms of the disease were muscle weakness and gait disturbance in 90.2% of patients and increased CPK in 9.8%. Calf pseudohypertrophy was seen in 60.9% of patients. Waddling gait was seen in 39% of patients. Gowers' sign was seen in 65.9% of patients. Contracture was observed in 12.2%. The prevalence of heart involvement showed that 7.3% of patients had heart involvement. Respiratory involvement was seen in 39% (Table 2).
| Parameters and Type/Determination | Percentage |
|---|---|
| Frequency of kinship | |
| Existence of parental kinship | 68.3 |
| Lack of parental kinship | 31.8 |
| Similar diseases | |
| Existence of a similar disease | 39 |
| Lack of a similar disease | 61 |
| Early symptoms of the disease | |
| Muscle weakness and gait disturbance | 90.2 |
| Increased CPK | 98 |
| Frequency distribution of leg pseudohypertrophy | |
| Positive | 60.9 |
| Negative | 39.1 |
| Waddling gait | |
| Positive | 39 |
| Negative | 61 |
| Frequency of Gowers' sign | |
| Positive | 65.9 |
| Negative | 34.9 |
| Contracture | |
| Yes | 12.2 |
| No | 87.8 |
| Cardiac involvement | |
| Yes | 7.3 |
| No | 92.7 |
| Respiratory involvement | |
| Yes | 34 |
| No | 66 |
Findings of Research Parameters
Regarding the early symptoms of the disease, there was muscle weakness progression in the form of independent walking in 32 out of 41 (78%) patients, dependent walking in 1 (2.4%) patient, and wheelchair dependence in 8 (19.6%) patients. The mean CPK level in patients was 9 442 U/L (Table 3).
| Variables | Values |
|---|---|
| Muscle weakness progression | |
| Independent walking | 78% |
| Dependent walking | 2.4% |
| Wheelchair dependence | 19.6% |
| CPK level (U/L) | |
| Mean | 9442 |
Results of Early Symptoms of the Disease
The genetic mutation study showed that among the genetically confirmed patients, 27.3% had SGCB mutation (LGMD R4), 18.2% had SGCA mutation (LGMD R3), 13.6% had SGCG mutation (LGMD R5), 9% had CAPN3 mutation (LGMD R1), 9% had ISPD mutations (FKRP), 9% had J2, 5.4% had 1B, 5.4% had 2B, and 5.4% had 2P (Table 4). The average CPK level differed significantly with mutation type (P = 0.013).
| Parameter | Type of Mutation | Percentage | CPK Level (U/L) | P-Value |
|---|---|---|---|---|
| Genetic mutation | SGCB | 27.3 | 9131 | 0.013 |
| SGCA | 18.2 | 9511 | ||
| SGCG | 13.6 | 8232 | ||
| CAPN3 | 9 | 9372 | ||
| J | 9 | 9435 | ||
| ISPD | 9 | 9297 | ||
| ISPD | 9 | 9553 | ||
| B1 | 5.4 | 9017 | ||
| B2 | 5.4 | 9921 | ||
| P2 | 5.4 | 9242 |
Relationship Between Type of Mutation and Creatine Phosphokinase Level
The relationship between CPK and pathological data was evaluated based on the type of mutation. There was no significant relationship between the CPK average level and regenerating fibers (%), degenerating fibers (%), and Lobulated fibers (%) (P > 0.05) (Table 5).
| Type of Mutation | Regenerating Fibers (%) | CPK Level | P-Value | Degenerating Fibers (%) | CPK Level | P-Value | Lobulated Fibers (%) | CPK Level | P-Value |
|---|---|---|---|---|---|---|---|---|---|
| SGCB | 4.7 | 9152 | 0.13 | 0.3 | 9124 | 0.09 | 15.8 | 9122 | 0.08 |
| SGCA | 2.3 | 9515 | 0.2 | 9492 | 3.0 | 9510 | |||
| SGCG | 3.5 | 8238 | 0.7 | 8432 | 0 | 8239 | |||
| CAPN3 | 2.1 | 9342 | 0.5 | 9355 | 3.3 | 9361 | |||
| J | 1.8 | 9407 | 0.1 | 9472 | 2.3 | 9409 | |||
| ISPD | 2.6 | 9214 | 0.4 | 9278 | 0.2 | 9294 | |||
| ISPD | 8.1 | 9523 | 0.0 | 9552 | 0.2 | 9541 | |||
| B1 | 2.6 | 9010 | 0.2 | 9033 | 21.6 | 9025 | |||
| B2 | 10.3 | 9924 | 0.7 | 9789 | 1.5 | 9917 | |||
| P2 | 7.5 | 9416 | 0.8 | 9471 | 0 | 9444 |
Investigating the Relationship Between Creatine Phosphokinase and Pathological Data Based on Mutation Types
5. Discussion
Similar large population-based studies have shown that 9.1% of the total muscular dystrophy identified is LGMD (23, 24). The present study showed that LGMD2D alpha sarcoglycanopathy was the most common type of LGMD. Contradictory results have been seen in other studies. Similar findings in several studies show that LGMD2A is the most common type of LGMD (9-11). A study conducted in Italy by Guglieri et al. showed that 84% of LGMD patients had the AR form, and LGMD2A was the most common LGMD (12). In other studies, calpainopathy was most common in different parts of the world (12, 13). However, in our study, calpainopathy was the fourth most common type of LGMD after alpha sarcoglycanopathy, beta sarcoglycanopathy, and gamma sarcoglycanopathy.
In a study carried out in Italy by Guglieri et al., after calpainopathy, dysferlinopathy was the second most common, and sarcoglycanopathies were the third most common LGMD (12). In a study carried out by Magri et al. in 2016 in Italy, LGMD2A and 2B were the most common subtypes of LGMD (17). The prevalence of dysferlinopathy is different in various studies. In our study, the prevalence of dysferlinopathy was 7.3%. In a consistent study, the prevalence of dysferlinopathy in the United Kingdom was reported to be 5.9% (11), while in Italy, it was 18.7% (12). However, as in the present study, the prevalence of dysferlinopathy was very low in the Netherlands (10).
In the present study, the prevalence of scoliosis was 7.3%, while in the study by van der Kooi et al. in the Netherlands, scoliosis was observed in 26.4% of patients (25). Also, in our study, the most common cases were alpha sarcoglycanopathy, followed by beta sarcoglycanopathy. The findings of Alavi et al. on 25 patients with sarcoglycanopathy in Iran showed that 14 patients had beta sarcoglycanopathy, 7 had gamma sarcoglycanopathy, 3 had delta sarcoglycanopathy, and 1 patient had alpha sarcoglycanopathy (26). Our study showed that among the patients studied, 27.3% had SGCB mutation (LGMD R4), 18.2% had SGCA mutation (LGMD R3), 13.6% had SGCG mutation (LGMD R5), 9% had CAPN3 (LGMD R1), and 9% had ISPD mutation (FKRP). Other studies have shown that the most common cases of LGMD in large and genetically heterogeneous populations are certain types such as LGMD R1 (CAPN3), LGMD R2 (DYSF), LGMD R3 (SGCA), and LGMD R4 (27-32).
In the present study, 28 out of 41 patients had sarcoglycanopathy. Parental kinship was present in 19 (67.9%) patients, and similar disease history was present in 11 (39.3%). Calf pseudohypertrophy was present in 60.9% of patients with sarcoglycanopathy, but in Alavi et al.'s study, all patients with sarcoglycanopathy had leg muscle pseudohypertrophy (26). In our study, cardiac involvement was present in 2 patients (7.3%). One patient had alpha sarcoglycanopathy, and one patient had gamma sarcoglycanopathy. In Alavi et al.'s study, there were cardiac abnormalities in six patients (24%); 5 patients had beta sarcoglycanopathy, and one patient had delta sarcoglycanopathy (26). In contrast, in the study by Urtasun in Spain, pseudohypertrophy of leg muscles or contracture was present in no patient (16).
According to previous studies, cardiac involvement is more common in beta sarcoglycanopathy than in other types of sarcoglycanopathies. Thus, the absence of cardiac involvement in patients with beta sarcoglycanopathies in our study is notable.
In the present study, contracture was present in 14.3% of patients with sarcoglycanopathy; 2 patients had alpha sarcoglycanopathy, one patient had beta sarcoglycanopathy, and one patient had gamma sarcoglycanopathy. In Alavi et al.'s study, 84% of patients had scoliosis in the late stages of the disease (26). Therefore, it seems that longer follow-ups are needed to observe the contracture.
Studies to offer new pharmacological methods considering biomarkers on various SGC mutations have shown that targeting sarcoglycan proteins and genes involved in dystrophy can be effective in reducing disease complications (7, 33-35). It is hoped that in the future, many problems of patients will be solved by offering new therapeutic solutions.
In the present study, SGCB and SGCA mutations were related to CPK levels. This finding has been confirmed in previous studies. Similar findings have shown that CPK changes in patients differ based on genetic mutations. The parallel process of reduction in CPK values in the mutated SGCB gene has shown a decrease in muscle, respiratory, and cardiac function (36). These findings indicate a possible genotype-phenotype correlation in this disease.
Based on the present findings, the level of CPK also showed a significant difference with types of mutations, as reported in other studies. In a study of patients with limb girdle muscular dystrophy type 2C, the average CPK was 10,300 (depending on the SGCG gene, it varied from 1,300 to 35,000 U/L) (37). In addition, in another study, the average CPK was 1154 in SGCB and 9000 - 15000 U/L in LGMD 2D (38, 39). An LGMD patient was evaluated with a CPK of 26,123 U/L. Neurodevelopmental disorders have been associated with a missense mutation in the LGMDR3 gene (40). These findings show the difference in CPK levels in different mutations, which was confirmed in the present study.
In general, physicians' failure to recognize the signs and symptoms of muscular dystrophy leads to unnecessary diagnostic measures. The study of the disease symptoms can be effective in recognizing and providing better therapeutic strategies.
One of the limitations is the lack of a patient registry system in the country, which causes no accurate information about diseases, including muscular dystrophy. If a national registry system is launched, most problems of the medical staff will be solved. Also, due to the fact that metabolic and genetic tests are not covered by insurance, performing these tests imposes heavy costs on parents. Health organizations in developing countries need to pay attention to this issue.
5.1. Conclusions
The prevalence of alpha and beta sarcoglycanopathy phenotypes in the study population showed that the severity of clinical involvement may be predicted by evaluating various mutations in the SGCB gene and the mentioned clinical symptoms.
