





1. Background
Primary ciliary dyskinesia (PCD) is predominantly inherited as an autosomal recessive disease. Very rarely, X-linked transmission (1) and autosomal dominant inheritance of PCD (2) have also been reported. Primary ciliary dyskinesia characteristics include chronic lung disease, rhinosinusitis, hearing impairment, and low fertility. Approximately 50% of patients with PCD exhibit a triad of symptoms known collectively as Kartagener syndrome. It has been reported that more than 40 mutated genes cause PCD, with known PCD-associated genetic variants detected in about 70% of PCD patients (3). Due to the low incidence rate of PCD and a lack of experienced and knowledgeable pediatricians who can provide accurate PCD diagnoses, the disease is frequently misdiagnosed and/or diagnosis is delayed. This situation is extremely unfavorable for children who are still in the early stage of PCD, as treatment during early-stage disease is critically important for slowing disease progression and improving patient outcomes and prognosis.
2. Objectives
The present study reviewed clinical characteristics, results of genetic testing (conducted via next-generation sequencing), PCD manifestations, and transmission electron microscopy (TEM) findings for 8 pediatric PCD patients admitted to Quanzhou Women’s and Children’s hospital in China in January 2019 to January 2022. The obtained results should enhance understanding of PCD.
3. Methods
3.1. Patients
Eight children diagnosed with PCD admitted to Quanzhou Women’s and Children’s hospital between January 2019 and January 2022 were included. A diagnosis of PCD was mainly based on clinical manifestations (at least two of the four key PCD features), TEM-detected structural ciliary defects, family history, and/or detection of known PCD-associated gene variants and novel gene variants (4). The four key PCD characteristics are as follows:
(1) Respiratory distress of term infants due to unknown cause
(2) Chronic wet cough starting before 6 months of age
(3) Chronic daily nasal congestion beginning before 6 months of age
(4) Abnormal left-right asymmetry of organs (5).
Informed consent was obtained from the parents and/or legal guardians of all the study participants. The study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of Quanzhou Women’s and Children’s hospital (No [2020]-7).
3.2. Methods
The clinical data were retrospectively analyzed and assessed. Variables of age, gender, onset age, age at diagnosis, clinical manifestations, computed tomography (CT) findings from sinus and lung scans, lung function test results, detected bacteria in patient sputa, detected gene mutations, and treatment and treatment outcomes were recorded.
3.3. Analysis of Genetic Mutations
Deoxyribonucleic acid (DNA) samples were extracted from peripheral blood leukocytes using standard genomic DNA purification methods. Whole-exome sequencing, bioinformatics analysis, and Sanger sequencing validation of detected sequence variants were performed. The pathogenicity analysis of detected gene variants was conducted, with results interpreted according to guidelines issued by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) (6). Sanger sequencing of gene regions corresponding to suspected variants was also conducted to verify sequence variants. By referring to the population frequency and pathogenic variation databases (Human Gene Mutation Database and ClinVar database, respectively), variants that were predicted to affect protein structure were identified using bioinformatics software, such as SIFT, PolyPhen2, and PROVEN.
3.4. Statistical Analysis
After data analysis via descriptive methods, categorial value-based results were expressed as percentages (%), and the results obtained via the analysis of non-parametric data were expressed as median and extreme values.
4. Results
4.1. Essential Patient Clinical Information
Of 8 children diagnosed with PCD at Quanzhou Women’s and Children’s hospital between January 2019 and January 2022, 5 cases received PCD diagnoses based on biallelic gene mutations combined with observed clinical manifestations, laboratory test results, and family history; nevertheless, 2 children were diagnosed based on results of mucosal biopsies performed 8 weeks after acute infection showing loss of inner dynein arm (IDA) and outer dynein arm (ODA) (IDA + ODA) (Figures 1 and 2), and 1 child was diagnosed based on observed clinical features consistent with Kartagener syndrome (Table 1 and Figure 3), which was considered to be PCD even if the previously mentioned criteria were not fulfilled completely. The parents and siblings of the 8 PCD patients lacked clinical PCD phenotypic characteristics. For the 8 PCD cases (2 males and 6 females), the median age at diagnosis was 5.75 years (range: 0.08 - 12 years).
 of biopsies from bronchial mucosa of a 10-year-old female primary ciliary dyskinesia (PCD) patient on admission (Case 2), showing lost integrity of cilia; detection of microtubular disorganization and partial loss of outer and inner dynein arms.")
Transmission electron microscopy (TEM) of biopsies from bronchial mucosa of a 10-year-old female primary ciliary dyskinesia (PCD) patient on admission (Case 2), showing lost integrity of cilia; detection of microtubular disorganization and partial loss of outer and inner dynein arms.
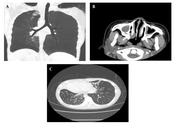
 and bronchoscopy image of a 10-year-old female primary ciliary dyskinesia (PCD) patient on admission (Case 2), showing peribronchial wall thickening and bronchiectasis (A) and thinned mucosa and sharpened tracheal ring (B).")
Chest computed tomography (CT) and bronchoscopy image of a 10-year-old female primary ciliary dyskinesia (PCD) patient on admission (Case 2), showing peribronchial wall thickening and bronchiectasis (A) and thinned mucosa and sharpened tracheal ring (B).
 image of a 12-year-old female primary ciliary dyskinesia (PCD) patient on admission (Case 3), showing dextrocardia and bronchial inflammation (A), rhinosinusitis (B), bilateral diffuse bronchiectasis and mucus plugging (C).")
Paranasal sinus and chest computed tomography (CT) image of a 12-year-old female primary ciliary dyskinesia (PCD) patient on admission (Case 3), showing dextrocardia and bronchial inflammation (A), rhinosinusitis (B), bilateral diffuse bronchiectasis and mucus plugging (C).
| No | Gender | Age at Diagnosis | Clinical Presentations | Radiological Manifestations | Sputum Pathogen | Pulmonary Function | Gene | TEM |
|---|---|---|---|---|---|---|---|---|
| 1 | Female | 8 y | Chronic wet cough/ nasosinusitis/ adenoid hypertrophy | Bronchiectasis | Pseudomonas aeruginosa | - | - | Loss of inner and outer dynein arms |
| 2 | Female | 10 y | Chronic wet cough/asthma/ nasosinusitis/ moderate malnutrition | Bronchiectasis | Klebsiella pneumoniae, Pseudomonas aeruginosa, Haemophilus influenzae | Mild obstructive dysfunction | Negative | Loss of inner and outer dynein arms |
| 3 | Female | 12 y | Chronic wet cough/ visceral inversion/ nasosinusitis | Bronchiectasis | Normal | - | - | - |
| 4 | Male | 1 mon | Postnatal respiratory distress/ visceral inversion | Atelectasis | Normal | - | CFAP300 | - |
| 5 | Female | 4 y | Chronic wet cough/ nasosinusitis/ gastroesophageal reflux/ adenoid hypertrophy/ moderate malnutrition | Inflammation | Normal | - | DNAH11 | - |
| 6 | Female | 6 y | Chronic wet cough/ nasosinusitis/ secretory otitis media | Atelectasis | Normal | Mild obstructive dysfunction | RSPH4A | - |
| 7 | Female | 5.58 y | Chronic wet cough/ nasosinusitis/ asthma/ secretory otitis media | Inflammation | Normal | Mild obstructive dysfunction | DNAH11 | - |
| 8 | Male | 1 mon | Postnatal respiratory distress/ visceral inversion/ intestinal malrotation with midgut torsion/ nasosinusitis | Bronchiectasis | Streptococcus pneumoniae | - | DNAH5 | - |
Clinical Characteristics of 8 Children with Primary Ciliary Dyskinesia
4.2. PCD Clinical Manifestations
Among the 8 PCD cases, 6 cases exhibited recurrent respiratory infections and sinusitis manifesting as cough, expectoration, nasal congestion, and purulent runny nose; however, 2 newborn cases born at term gestation exhibited shortness of breath, respiratory distress, and excessive sputum production after birth. With disease progression, all cases exhibited recurrent cough and expectoration, 1 case developed recurrent runny nose, and 3 cases exhibited recurrent wheezing (2, 5, and 3 episodes) that were all alleviated by anti-infection and nebulization treatments. In addition, 2 cases were diagnosed with asthma, with wet rales noted in 7 cases and wheezing rales noted in 2 cases. Other PCD manifestations were also observed, with 3 cases diagnosed with total visceral inversion, 2 cases complicated with secretory otitis media, 2 cases complicated with adenoid hypertrophy, 1 case complicated with gastroesophageal reflux, 1 case with intestinal malrotation and midgut torsion, and 2 cases with moderate malnutrition.
4.3. Examination Findings and Laboratory Test Results
Among the 8 cases, examination findings included bronchiectasis (4 cases), diffuse bronchiectasis (3 cases), localized bronchiectasis (1 case), bilateral upper lobe bronchiectasis (3 cases), middle-lobe bronchiectasis (2 cases), lower-lobe bronchiectasis (2 cases), bronchial wall thickening (3 cases), and rhinosinusitis that was detected via paranasal sinus CT scans (7 cases). Microbiological testing of bronchial lavage fluid samples obtained during bronchoscopy performed on 6 children detected three bacterial species in the specimen from 1 child (i.e., Pseudomonas aeruginosa, Haemophilus influenzae, and Klebsiella pneumonia) and single bacterial species in specimens collected from two other patients, including Pseudomonas aeruginosa (1 case) and Streptococcus pneumoniae (1 case). The sputum culture results are related to the first sputum culture of the patients. Three children completed pulmonary function testing that revealed mild obstructive dysfunction (7) (3/3) and one positive bronchodilation test result (1/3).
4.4. Genetic Characteristics
Six children completed whole-exome sequencing testing. The results revealed that 1 case was negative for known variants; nevertheless, 5 cases were positive for known PCD-associated variants, with 1 case shown to have a homozygous variation and 4 cases shown to have compound heterozygous variants. Among the 4 cases with compound heterozygous variants, 2 cases possessed DNAH11 mutations, 1 case possessed a DNAH5 mutation, and 1 case possessed an RSPH4A mutation. Meanwhile, 1 case was homozygous for a CFAP300 variant; nevertheless, 8 novel variants were detected in 5 cases that included 3 DNAH11 variants (c.5460 + 5G > C, c.11749_11752delGTTA, and c.5822G > C), 2 DNAH5 variants (c.4314delT and c.877dupA), 2 RSPH4A variants (c.1774_1775delTT and c.1949A > G), and 1 CFAP300 variant (c.603delG). Novel variants included a splice-site variant and nonsense, missense, and frameshift variants that have not been previously reported (Table 2).
| No | Patient No | Gene | Region | Nucleotide Change | Amino Acid Change | Genotype | Variant Type | ACMG/AMP Pathogenicity | Reported/Novel |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Patient 4 | CFAP300 | Exon 5 | c.603delG | p.V202* | Homozygous | Nonsense | Likely pathogenic | Novel |
| 2 | Patient 5 | DNAH11 | IVS17 | c.3426 - 1G > A | - | Compound heterozygous | Splice variant | Pathogenic | Reported |
| 3 | Patient 5 | DNAH11 | IVS31 | c.5460 + 5G > C | - | Splice variant | VUS | Novel | |
| 4 | Patient 6 | RSPH4A | Exon 4 | c.1774_1775delTT | p.L592fs*5 | Compound heterozygous | Frameshift variant | Likely pathogenic | Novel |
| 5 | Patient 6 | RSPH4A | Exon 6 | c.1949A > G | p.H650R | Missense | VUS | Novel | |
| 6 | Patient 7 | DNAH11 | Exon 72 | c.11749_11752delGTTA | p.V3917fs*20 | Compound heterozygous | Frameshift variant | Likely pathogenic | Novel |
| 7 | Patient 7 | DNAH11 | Exon 34 | c.5822G > C | p.W1941S | Missense | VUS | Novel | |
| 8 | Patient 8 | DNAH5 | Exon 27 | c.4314delT | p.N1438fs*10 | Compound heterozygous | Frameshift variant | Likely pathogenic | Novel |
| 9 | Patient 8 | DNAH5 | Exon 7 | c.877dupA | p.R293fs*6 | Frameshift variant | Likely pathogenic | Novel |
Gene Variations in 5 Children with Primary Ciliary Dyskinesia
4.5. PCD Treatments and Outcomes
In this study, routine treatment of PCD included antibiotics, anti-inflammatory administration, airway clearance, and inhaled steroids. The airway clearance technique was necessary for effective PCD long-term management. Eight children were treated with inhaled steroids to alleviate airway inflammation and antibiotics that included cefoperazone/sulbactam, amoxicillin-clavulanate, piperacillin/tazobactam, and meropenem to combat bacterial infections. Six children also received anti-inflammatory oral azithromycin treatment, and 1 child received intravenous immunoglobulin (IVIG) for her long acute exacerbation duration. In addition, case 4 and case 8 underwent mechanically-assisted ventilation for neonatal respiratory distress.
After receiving acute-phase treatments by children, PCD symptoms of all children were relieved to varying degrees, with the stabilization of symptoms achieved in 6 cases. For the 2 remaining cases, 1 case (case 8) still suffered from recurrent cough and expectoration after treatment; however, the other case (case 4) was referred to another hospital for further treatment due to continued ventilator dependence. Five children (cases 1, 2, 5, 6, and 7) received training on how to clear mucus from their lungs using several techniques, such as postural drainage, percussion, cough control, and active circulatory breathing.
5. Discussion
Primary ciliary dyskinesia is a rare respiratory disease in children, with an incidence rate of 1:10000 - 1:20000 (8). The disease often occurs early in life, with many infants, even full-term infants, exhibiting respiratory distress at birth (9). Nonetheless, only 73% of children with PCD have been reported to exhibit neonatal respiratory symptoms (10), thereby indicating that although PCD starts early, it is not generally a lethal disorder. Prognosis can be improved if PCD is diagnosed early so that appropriate protective measures can be implemented to delay the occurrence of bronchiectasis and prevent recurrent respiratory tract infections. Nevertheless, diagnosis is often delayed, as indicated in one early study conducted in the United Kingdom that reported an average age of PCD diagnosis at 4.4 years (11). In a larger study conducted more recently on 1,009 PCD patients in 26 European countries and China, median ages at diagnosis of 5.3 years and 9.16 ± 3.67 years were reported, respectively (12). More recently, a study conducted at Beijing Children’s hospital in China reported a median age at diagnosis of 7.0 years (2 months to 14 years) for 75 pediatric PCD patients admitted between 2012 to 2019 (13). In the current study, the median age of diagnosis was 5.75 years as an indicator of earlier PCD diagnosis in China during the past 3 years.
The accurate diagnosis of PCD, a complex genetic heterogeneous disease, depends on the collection of medical information from a variety of sources (e.g., medical history, symptoms, examination findings, TEM analysis of ciliary ultrastructure analysis, and genetic testing) since no gold standard PCD diagnostic assay yet exists. The most useful and convenient first step for PCD diagnosis is to carefully examine the patient’s medical history and symptoms and compare these clinical features to four key PCD clinical features outlined in the American Thoracic Society clinical guidelines (14). By basing a diagnosis on a positive match between patient clinical features and at least two of the four key clinical features outlined in the guidelines, PCD diagnostic specificity and sensitivity might be improved.
Importantly, expectoration in PCD patients is more pronounced than in other respiratory disorders, with symptoms in PCD cases almost always beginning at a very young age (most often shortly after birth) and improving (but never disappearing completely) under good weather conditions and after the administration of standard anti-asthma or anti-allergy treatments. Primary ciliary dyskinesia-related symptoms observed in this study included wet cough (100%, 8/8), neonatal respiratory distress (25%, 2/8), bronchiectasis (50%, 4/8), nasal congestion and runny nose (87.5%, 7/8), and otitis media (25%, 2/8). Moreover, PCD symptoms were accompanied by several other disorders, such as asthma, gastroesophageal reflux (15), esophageal or extrahepatic biliary atresia, intestinal malrotation, and/or abnormal spleen or kidney development, with asthma, gastroesophageal reflux, and intestinal malrotation manifestations noted in these patients.
Although not all PCD-associated genes are known at present, PCD-related gene variants have been detected in about 65 - 70% of PCD cases. Therefore, a negative genetic test result cannot be used to completely rule out PCD as a diagnosis. However, as the price of genetic testing continues to fall, more variants will likely be discovered that will further broaden the known PCD spectrum of gene variants and, therefore, increase the proportion of cases that can be detected using this method. For example, genetic testing results for case 2 were normal (no PCD-associated gene variant); nonetheless, TEM analysis of ciliary ultrastructure after 8 weeks of infection revealed an absence of IDA and ODA as the basis for a PCD diagnosis for this patient. Nevertheless, until genetic testing can detect PCD in 30 - 35% of suspected patients with negative genetic test results, the ultrastructural examination of cilia will remain an important tool for PCD diagnosis. Meanwhile, TEM-associated technical difficulties in capturing informative ciliary ultrastructure defects in young children, poor compliance of tissue sampling in pediatric patients and parents, and potential secondary changes in mucosa and cilia ultrastructure can greatly impact TEM results (16, 17). In the group of PCD patients studied in this investigation, only 2 children underwent successful ciliary biopsy; nevertheless, 2 newborns were diagnosed early through genetic examination alone since they were unable to produce specimens for TEM analysis.
To date, the relationship between PCD genotype and phenotype has not been fully clarified. One study reported that patients with CCDC39 or CCDC40 mutations had worse lung function and growth indices than those with ODA defects and DNAH5 mutations, respectively (18). With the rapid development of molecular biological tools and gene sequencing platforms, more and more PCD pathogenic genes have been discovered that have enhanced our understanding of the PCD genotype-phenotype relationship.
The genetic spectrum of PCD variants varies among populations in different countries (19). Due to the fact that ultrastructural abnormalities are easy to identify, variants of DNAI1 and DNAH5 genes in PCD patients were the first to be characterized, with high prevalence rates found for DNAH5 (15 - 21%) and DNAI1 (9%) variants in these patients. Subsequently, Kazuhiko (20) reported that if mutations were not detected within mutational hot spots of DNAI1 and DNAH5, whole-exome sequencing could be performed to attempt to detect variants of those genes. Notably, variants of DNAH5, a gene encoding a protein involved in the assembly of the ODA, can cause visceral transposition, a characteristic observed in case 8 at birth that was consistent with a positive test result for a DNAH5 variant. However, the cilia of patients with known PCD-associated genotypic variants of DNAH11, CCDC164, CCDC65, and OFD1 genes have been reported to possess normal ultrastructural characteristics. Nevertheless, although DNAH11 variants were detected in 2 cases in the present study, their associations with ciliary dysfunction could not be assessed via TEM since parental approval for mucosal biopsy was not provided.
RSPH4A is involved in encoding the radial spoke head that plays a role in upstream signal transduction for the activation of ciliary motion involving the central microtubule and dynein arm. Immunofluorescence data and cryo-electron tomography data suggested that the spoke head and the neck/arch were disrupted in the absence of RSPH4A in the mouse motile cilia (21). Due to the fact that no central microtubules exist within the cilia of the embryonic node (i.e., a structure that determines left- or right-sidedness of the viscera), the mutations of the gene encoding the radial spoke do not cause visceral transposition, as exemplified by an RSPH4A founder mutation (c.921+3_6delAAGT) that is viewed as a pathogenic PCD variant that does not cause lateral defects (22). This RSPH4A mutation disrupts the normal ciliary planar beating mode, resulting in circular ciliary motion (23). In this study, case 5 exhibited a relatively later onset of atelectasis, nasosinusitis, and secretory otitis media without visceral transposition, as consistent with known phenotypic characteristics associated with RSPH4A variants.
Children with variants of CFAP300 (also known as C11orf70) share a consistent PCD phenotype beginning early in life that includes laterality defects and immotile respiratory cilia due to loss of both IDA and ODA (24, 25). Notably, in this study, case 4 exhibited postnatal respiratory distress and visceral inversion that were consistent with the phenotype associated with this genotype. Using SIFT and PolyPhen2 protein destructive prediction software, the current study analyzed the effects of this c.603delG mutation and verified that this mutation might alter protein structure, which could lead to loss of protein function.
Due to the fact that no specific treatment for PCD has existed until recently, it has been important to diagnose PCD as early as possible so that treatment administration and long-term management can be started early in life to improve long-term prognosis. The current recommended treatment resembles treatments used to alleviate bronchiectasis caused by other disorders (e.g., cystic fibrosis) that delay disease progression by improving or maintaining lung function while preventing chronic lung injury, although regular airway cleaning and proactive administration of treatments to prevent airway infections. Importantly, the results of the first multinational randomized controlled trial on PCD drug treatment revealed that 6-month azithromycin maintenance treatment was well tolerated and halved the incidence of acute respiratory exacerbations (26). The results of the aforementioned study have led to the use of azithromycin to treat pediatric PCD patients who experience frequent acute exacerbations in order to minimize the use of additional antibiotics while preventing irreversible lung injury. In the current study, disease progression was prevented in patients who received oral azithromycin for 1 to 6 months without triggering obvious side effects.
This research study had limitations. Firstly, the data were obtained from a single center, which might limit the generalizability of the findings. Secondly, the sample size was small, and further research is needed to confirm the obtained findings and explore potential differences in clinical manifestations between different subtypes of PCD. Moreover, nasal nitric oxide testing or immunofluorescence, the major diagnostic tools, were not used for these patients. Lung function tests, hearing tests, and mucosal and ciliary biopsies were not completed in all patients, with missing data limiting the analysis of some variables. Nonetheless, the present study’s results highlight the importance of early PCD diagnosis and case treatment management as conclusions that await confirmation through additional investigations based on larger, more diverse patient cohorts.
5.1. Conclusions
Clinical PCD manifestations, such as recurrent cough, expectoration, purulent discharge, bronchiectasis, and visceral inversion, and laboratory test results obtained using TEM and gene sequencing are important for achieving early PCD diagnosis. In addition, genotypes discovered in this study will expand the PCD genotypic spectrum.
