



1. Introduction
Hunter syndrome or mucopolysaccharidosis-II (MPS-II) is the clinical sequel of glycosaminoglycans dermatan and heparan sulfate accumulation in tissues due to iduronate-2-sulfatase deficiency (1). Hunter syndrome occurs almost exclusively in males and has been reported at a rate of about 1 in 170,000 male births (2). All patients have somatic involvement, including facial dysmorphism, enlarged liver and spleen, stiff joints and contractures, valvular heart disease, and upper airway obstruction (2). Mucopolysaccharidosis-II is diagnosed by identifying deficiency in iduronate 2-sulfatase enzyme activity in white cells, fibroblasts, or plasma in a male proband in the presence of the normal activity of at least one other sulfatase. Detection of a hemizygous pathogenic variant in IDS confirms the diagnosis in a male proband (3).
According to the literature, 660 mutations in the IDS gene have been identified (4), 65% of which are caused by single-nucleotide changes and splicing variants and 17% by small (i.e., intra-exon) deletions and insertions. These mutations can be detected by IDS gene sequencing (3). About 9% of mutations are caused by intragenic deletions or duplications. These mutations can be detected by gene-targeted deletion/duplication analysis methods, such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications (3). The remaining 9% of mutations are caused by complex rearrangements resulting from recombination with the pseudo-gene IDSP1 or other processes. The assay uses multiple molecular methods to verify and map the rearrangement breakpoints (e.g., may require sequencing, SNP analysis, gene-targeted deletion/duplication analysis, and chromosomal microarray) (5).
In this article, we present the process of detecting the mutation in a patient diagnosed with MPS-II, in which the pathogenic variant could not be detected by IDS gene sequencing.
2. Case Presentation
Our patient, who had an uneventful fetal period and delivery history, was operated on for a bilateral inguinal hernia in the following months. He was referred to our center when he was 1 year old due to frequent recurrent upper respiratory tract infections and rough facial appearance. On physical examination, rough facial appearance, eczema on both cheeks, 3 cm hepatomegaly and splenomegaly, large-looking hands, short fingers, and gingival hypertrophy were present. Arylsulfatase B [4.7 nmol/mg/hour (reference: > 10)] was found to be low in the MPS enzyme screening, which can be measured at a limited level. The iduronate-2-sulfatase level could not be measured. Galsulfase treatment was started for the patient. Meanwhile, the ARSB gene was studied by the next-generation sequencing (Miseq-Illumina) method covering the coding regions of the gene. When no mutation was detected in the ARSB gene, galsulfase treatment was discontinued for 4 weeks. Then, all MPS enzyme levels were rechecked. This time, while arylsulfatase B and other MPS enzymes were within normal limits, only iduronate 2-sulfatase level [< 0.1 nmol/mg.hour (reference > 7.5)] was found to be low. After the results were confirmed in two more different centers, IDS gene analysis was sent, and idursulfase treatment was started. No mutation was detected in the IDS gene by the next-generation sequencing (Miseq-Illumina) method covering the coding regions of the gene. Therefore, we tried to detect a mutation in the IDS gene by RNA sequencing.
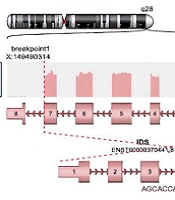
Total RNA from the peripheral blood of the patient was isolated using the QIAamp RNA Blood Mini Kit (Qiagen, Germany). To remove residual genomic DNA, the sample was treated with RNase-free DNase I (New England BioLabs). Hybridization-based rRNA depletion kit (MGIEasy rRNA Depletion Kit) was used to remove the ribosomal RNA population. A total of 200 ng of depleted RNA sample was used for sample preparation with MGIEasy RNA Library Prep Kit. The sample library was pooled along with other samples and was sequenced paired-end (2 × 150 bp) on MGI DNBSeq G400, targeting 12.5 Gb raw data per sample. Obtained short reads were aligned to the hg38 reference genome using the STAR alignment tool. For the detection of gene fusions, the Arriba algorithm was used (6). Findings suggesting an inversion in the IDS gene and a fusion between the EOLA1 gene located next to this gene were detected by this method (Figure 1).
 gene and a fusion between the adjacent <i>EOLA1</i> gene")
Inversion in the iduronate 2-sulfatase (IDS) gene and a fusion between the adjacent EOLA1 gene
3. Discussion
The MPS II is caused by mutations in the IDS gene. This gene consists of nine exons, its locus contains 24 kb and is located on the long arm of the X chromosome (Xq28) (7). The IDS gene is most expressed in the brain and adrenal glands and in 21 more tissues (8). Nucleotide insertions, deletions (especially those causing frameshifts during translation), major rearrangements, and nonsense mutations are harmful (7). The spectrum of DNA variation in the human genome comprises small base changes (substitutions), insertions and deletions of DNA, large genomic deletions of exons or whole genes, and rearrangements, such as inversions and translocations (9).
Casas-Alba et al. stated that the results could be obtained by RNA sequencing, especially in splicing mutations (synonymous, splice site, or intronic mutations) and regulatory DNA mutations (promoter, enhancer, and others) that cannot be detected by exome sequencing (10). Due to the high genetic heterogeneity of the Hunter syndrome, no high-frequency mutations have been detected so far. It is known that almost half of the variants reported in the IDS gene are insertions/deletions (gross or small), complex re-arrangements, or splicing variants (11). With a targeted gene sequencing approach with NGS methodology, it is impossible to detect such variations by only coding the regions of the gene sequenced. When the clinical features are solid but targeted gene panels fail to reveal underlying mutations, it is recommended to use second-tier tests, such as genome sequencing, exome sequencing, or transcriptome analysis (10). The choice among recommended alternatives may depend on availability, price, experience with analysis pathways, and clinical expectations. In the case of Hunter syndrome, it is known that splicing variants on the IDS gene can cause the disease (12). RNA-based approaches (whole transcriptome sequencing or targeted analysis on the cDNA samples) are the best ways to study splicing variants and elucidate their effects. Moreover, it is possible to analyze fusion events after whole transcriptome sequencing. Therefore, whole transcriptome sequencing was the best alternative for revealing any splicing and/or fusion events in this case.
In our case, we could not detect a mutation in the IDS gene using next-generation sequencing (Miseq-Illumina), and we could not find a center to run IDS gene analysis using the MLPA method. Therefore, we tried to detect the mutation in the IDS gene by RNA sequencing. We detected complex rearrangements with RNA sequencing, suggesting an inversion in the IDS gene and a fusion between the adjacent EOLA1 gene. As a result, we confirmed the diagnosis with the molecular genetic method, and we were able to provide genetic counseling to the family. The main features of the next-generation sequencing of DNA and RNA sequencing are summarized in Table 1.
| Next-Generation DNA Sequencing | RNA Sequencing |
|---|---|
| NGS can sequence entire genomes or be constrained to specific areas of interest, including all 22000 coding genes (a whole exome) or small numbers of individual genes (9). | RNA-seq works by sequencing every RNA molecule in a sample and measuring the expression of genes by counting the number of times its transcripts have been sequenced (13). |
| The main advantage of NGS technologies is to avoid the bacterial cloning of DNA fragments and electrophoretic separation of sequencing products (14). | Disease-relevant tissue is the best sample for RNA-seq because gene expression and splicing patterns are unique to each tissue and cell type (13). |
| Limitations of NGS include the analytic sensitivity of mutation detection, areas of the genome that are difficult to sequence or analyze, limitations in the knowledge of how to interpret novel or rare mutations, limited ability to detect structural gene variation and copy number variation, and integration of genomic information (15). | Single-gene RNA sequencing can elucidate the role of the candidate genetic variants of uncertain significance (VUS) by combining reverse transcriptase to create cDNA and RT-PCR to measure RNA (10). |
| The main disadvantage is their short read length. Misassembly and breaks may occur because genomes contain many repetitive sequences that are slightly longer than the length read by NGS (14). | Performing RNA sequencing (RNA-seq) of the entire transcriptome in a single run can facilitate the detection of aberrant expression, aberrant splicing, and mono-allelic expression (10). |
| Large structural variations are difficult to find and characterize, while single nucleotide variations and short indices can be accurately detected with short reads (14). | The main challenges of RNA-seq are the accessibility of appropriate tissues for mRNA extraction, the large amount of sequencing required to detect changes, and the necessity of large control cohorts (10). |
Features of Next-Generation Sequencing and RNA Sequencing
In diagnosing hereditary metabolic diseases, it is necessary to reach the diagnosis with biochemical analyses in light of clinical findings and confirm the diagnosis with molecular genetic analysis. As mentioned above, we could not confirm the initial diagnosis made by biochemical analysis with genetic analysis. Therefore, we reached the correct diagnosis from the wrong one. In some diseases diagnosed with clinical and biochemical methods, mutations cannot be detected even with advanced genetic methods, such as next-generation sequencing. In these cases, we emphasize that mutations should be investigated using different methods, such as RNA sequencing.
