1. Background
Carcinoid tumors, first described by Langhans in 1867, are among neuroendocrine neoplasms that can rarely become malignant (1). These tumors are usually located in the gastrointestinal tract (2). Although primary appendiceal neoplasms are rarely seen, the appendix is the most common site of carcinoid tumors (3). Carcinoids constitute over 32 - 57% of all appendiceal neoplasms (4). The second and third decades of life are the most common periods when carcinoid tumors are detected (5). Carcinoid tumors of the appendix do not have specific clinical presentations and are usually detected incidentally during the histopathological analysis of appendectomy specimens resected due to acute appendicitis.
Regarding the surgical management of appendiceal carcinoids, appendectomy is generally sufficient for sub-centimeter tumors, whereas a broader surgical approach, such as hemicolectomy, may be required for tumors larger than 2 cm. However, the surgical removal of tumors between 1 and 2 cm is a controversial issue (3). Therapeutic and follow-up protocols suggested for these tumors are mostly based on retrospective and relatively small-scale studies, probably due to their low incidence (5-7). Hence, it is important to enhance our knowledge to be able to appropriately manage appendiceal carcinoids, particularly in children.
2. Objectives
In this study, we aimed to evaluate the clinicopathological features and long-term outcomes of appendiceal carcinoid tumors in pediatric patients.
3. Methods
The data of 1562 patients under the age of 18 years who underwent surgery due to acute appendicitis from January 2014 to September 2022 were retrospectively analyzed. Patients diagnosed with appendiceal carcinoid tumors confirmed by histopathological analysis were included in the study. Studies with retrospective designs have limitations, such as reliance on archived medical records. In this study, potential limitations such as missing data or loss to follow-up were minimized by careful reviewing of available records and contacting patients for follow-up. Age, gender, preoperative clinical findings, surgical data, and histopathological records were collected. The patients diagnosed with carcinoid tumors were postoperatively followed up with annual abdominal ultrasonography (US) and 68Ga-DOTATATE uptake on positron emission tomography/computed tomography (PET/CT). The starting point of the follow-up period was considered the date of diagnosis. The findings obtained were compared with previous studies in the literature.
The data were analyzed using the Statistical Package for Social Sciences (SPSS) for Windows version 22.0 (IBM, Armonk, NY). Descriptive indices included mean and standard deviation (SD) for parameters with normal distribution and median and range for parameters with non-normal distribution.
The ethics approval code (ESH/GOEK 2022/8, 21.12.2022) was obtained from the University of Health Science, Eskisehir City Hospital.
4. Results
A total of 10 (0.64%) appendiceal carcinoid tumors were found after conducting 1562 appendectomies. The tumors belonged to 4 (40%) boys and 6 (60%) girls, with a mean age of 12.6 years (range: 8 - 17 years). In all patients, preoperative clinical findings were compatible with the acute abdomen syndrome. None of the patients had symptoms suggesting carcinoid syndrome, such as palpitations, dyspnea, hypertension, diarrhea, and flushing.
Regarding surgical procedures, open appendectomy and laparoscopic appendectomy were performed in seven and three patients, respectively. In the pathological evaluation, tumor localization was at the tip of the appendix in nine patients, whereas one tumor was located in the middle part of the organ. Most tumors (n = 7, 70%) were smaller than 1 cm, and 3 tumors were 1 - 2 centimeters in size. The depth of tumor penetration reached the serosa in eight patients and the mesoappendix in the remaining 2 patients. In patients with tumors larger than 1 cm, the depth of invasion was limited to the mesoappendix in two patients and submucosa in one patient. Surgical margins were intact in all specimens. The histologic features of acute appendicitis were seen in 8 (80%) out of 10 patients. In terms of histopathology, the tumor type was well-differentiated in all patients.
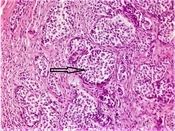
Hematoxylin and eosin-stained tumor sections showed small, narrow islets of the cytoplasm and cord-like structures around the nucleus (Figure 1). Immunohistochemistry revealed that the tumoral cells were positive for neuroendocrine cellular components, such as chromogranin A, synaptophysin (Figure 2), and neuron-specific enolase (NSE). There was no evidence of lympho-vascular or perineural invasion in patients. The Ki-67 proliferation index was positive in less than 2% of cells in nine (90%) of the cases and 4% of cells in one (10%) patient. In nine patients, the mitotic activity was found to be less than 2 in 10 high-power fields (HPF). Demographic data of patients, histopathological findings of tumors, and follow-up durations have been summarized in Table 1.
")
Figure 1.
The tumor appears to be composed of monotonous cells consisting of round nuclei with a fine-dotted chromatin network (H&E, 400x)
")
Figure 2.
Immunohistochemical staining showed diffuse strong positivity for synaptophysin, a neuroendocrine cellular marker, in the tumor (IHC, 100x)
Table 1.
Demographic and Histopathologic Findings and Follow-up Durations of the Patients (n = 10)
| Patient No. | Age (y) | Gender | Localization | Depth of Invasion | Size (mm) | Ki67 (%) | Follow-up (mo) |
|---|---|---|---|---|---|---|---|
| 1 | 10 | M | Tip | Submucosa | 10 | < 2 | 84 |
| 2 | 14 | F | Tip | Submucosa | 3 | < 2 | 32 |
| 3 | 14 | F | Tip | Submucosa | 2 | < 2 | 54 |
| 4 | 14 | M | Tip | Submucosa | 3 | < 2 | 72 |
| 5 | 17 | F | Middle | Mesoappendix | 11 | < 2 | 12 |
| 6 | 10 | M | Tip | Submucosa | 5 | < 2 | 44 |
| 7 | 10 | F | Tip | Mesoappendix | 12 | 4 | 24 |
| 8 | 12 | F | Tip | Submucosa | 5 | < 2 | 32 |
| 9 | 8 | F | Tip | Submucosa | 1 | < 2 | 42 |
| 10 | 17 | M | Tip | Submucosa | 6 | < 2 | 28 |
Abbreviations: Y, year; mm, millimeter; M: male; F, female.
The patients diagnosed with carcinoid tumors were followed up for an average of 42.4 months (ranging from 12 to 84 months). Abdominal ultrasounds and PET/CT scans were performed every 6 months in the first year and then annually to monitor recurrence. No regional or distant recurrences were observed in the follow-up, and no further surgical procedures, such as right hemicolectomy, were required. No mortality was observed during the follow-up.
5. Discussion
Contrary to adults, primary appendiceal neoplasms are extremely rare in children. The incidence of appendiceal carcinoid tumors has been reported to be 0.2 - 0.5% of all appendectomy specimens (6). In our study, the frequency of carcinoid tumors located within the appendix was calculated as 0.64%, which was in accordance with the literature. The mean age of children with carcinoid tumors is between 12 and 13 years old, and these tumors are more commonly seen in girls than in boys (1, 7, 8). Similarly, in our cohort, the mean age was observed to be 12.6 years, with a slightly female predominance.
There are no specific clinical and radiological findings for the diagnosis of appendiceal carcinoid tumors preoperatively (5). Carcinoid tumors of the appendix mostly present with nonspecific acute abdominal findings and are diagnosed incidentally in histopathological examination (5). In our study, none of the patients received a suspicion of carcinoid tumor during the preoperative work-up, and the diagnosis of this condition was established by the histopathological examination of the specimens obtained by appendectomy due to acute appendicitis. Carcinoid syndrome, which is characterized by skin flushing, bronchoconstriction, diarrhea, peripheral vasomotor symptoms, and right-sided heart valve fibrosis, may develop when vasoactive mediators (e.g., serotonin and histamine) are systemically released into circulation by the carcinoid tumor (9). This syndrome occurs in less than 2% of patients with appendiceal carcinoids (10). None of our patients presented with the symptoms of carcinoid syndrome.
In children, most carcinoid tumors are less than 2 cm in size and are located at the tip of the appendix in 75% of the cases (5, 11). This can explain their nonspecific clinical picture and radiological signature. In the present case series, all tumors were smaller than 2 cm, and nine of the ten tumors were located at the tip of the organ.
Simple appendectomy has been reported to be curative in most patients with carcinoid tumors (5). Some experts, however, advocate a more aggressive approach and recommend right hemicolectomy, especially for tumors located proximal to the appendix, high-grade malignant carcinoids, and carcinoids with a high mitotic index (5, 12). In our study, all tumors were small, and none of them were located at the base of the appendix. The resection margins were also reported to be free of cancer cells in histopathological analyses. Additionally, none of our patients required further surgical procedures during the postoperative follow-up period.
The cell proliferation rate is an important prognostic factor for neuroendocrine tumors (13). Therefore, proliferation markers, particularly Ki-67 (an indicator of mitotic activity), have become increasingly important in the evaluation of neuroendocrine tumors (NETs) (14). The World Health Organization (WHO) and the European Neuroendocrine Tumor Society (ENETS) have categorized NETs into three groups based on the grade, Ki-67 proliferation index, and mitotic activity. The presence of fewer than two mitoses and a Ki-67 proliferation index of < 2% in 10 HPFs are the features of “low-grade NET (Class 1)”. If there are 2 - 20 mitoses and a Ki-67 proliferation index of 3 - 20%, “intermediate NET (Class 2)" is diagnosed, and more than 20 mitoses and a Ki67 proliferation index greater than 20% are indicative of “high-grade NET (Class 3)” (15). In 9 out of our 10 patients, there were less than 2 mitoses per 10 HPFs with a Ki67 proliferation index of below 2%. In one patient, 2 mitoses were observed in 10 HPFs, and the Ki67 proliferation index was 4%. The latter patient was followed up for 24 months with annual abdominal ultrasonography and gallium-68 PET-CT scanning, and no recurrence was detected.
Although NETs are usually sporadic, some familial syndromes, including multiple endocrine neoplasia types I and II, von Hippel Lindau syndrome, and neurofibromatosis type I, can rarely be associated with these tumors (15). No genetic anomaly or additional disease was detected in our patients. Additionally, these tumors may be multifocal or associated with gastrointestinal stromal tumors. Therefore, imaging modalities such as computed tomography (CT), magnetic resonance imaging (MRI), ultrasonography, endoscopy, and functional imaging should be used during follow-up (16). Radio-labeled somatostatin analogs are valuable diagnostic tools for both diagnosis and treatment of NETs due to frequent tumoral overexpression of somatostatin receptors (17). The PET/CT imaging technique incorporated with radiolabeled somatostatin analogs (68Ga-DOTATATE PET/CT) is used as a new gold standard (18), which is superior to most imaging techniques due to low exposure to radiation, low toxicity, fast application/clean-up time, and low cost-effectiveness (17). Previous studies have suggested that this functional imaging modality should be used as a basic screening tool in adult and pediatric patients during follow-up and as a reliable method for optimizing therapeutic regimens for pediatric patients (18, 19). Abdominal ultrasound and PET-CT were performed for our patients every 6 months in the first year, and then annually. No recurrence was observed in these patients, and no additional surgical procedure (such as right hemicolectomy) was required. In case of suspicion of distant metastasis or synchronous gastrointestinal cancer, endoscopic examination of the gastrointestinal tract is recommended to identify the primary tumor and exclude accompanying malignancies (20). Since none of our patients had suspected metastasis or synchronous cancer, evidenced by imaging methods, none of them needed endoscopy.
Prognosis in appendiceal carcinoid tumors is better than in other midgut carcinoids (21), with the 5-year survival rate being 92% when the disease is limited to the appendix, 82% in cases with locoregional disease, and 31% in patients with distant metastases (21, 22). The patients in our cohort were routinely directed to the medical oncology department. No synchronous tumors, disease recurrence, metastases, or mortality were detected in our patients during the follow-up.
5.1. Limitations
One of the limitations of this study was its single-center and retrospective design. Also, post-adolescent follow-up should be supervised by adult surgeons, and then the results should be presented in another paper.
5.2. Conclusions
Childhood appendiceal carcinoid tumors are mostly detected incidentally following an appendectomy. For this reason, after the histopathological diagnosis of this tumor, careful follow-up, thorough examinations, and advanced surgical and medical treatments should be considered when necessary. Although survival is good, the possibility of developing colorectal malignancy should not be ignored during follow-up.