1. Introduction
Mutations in the KDM5C gene can cause Claes Jensen syndrome, an X-linked intellectual disability (ID). This condition typically presents in early childhood and is characterized by impairments in cognitive processes and an intelligence quotient (IQ) below 70 (1-3). The Lysine Demethylase 5C gene (KDM5C), located at Xp11.22, is one of the most significant genes causing moderate to severe ID in both syndromic and non-syndromic types of ID (4). To date, more than 35 mutations in KDM5C have been identified, which alter epigenetic regulation by removing di- and tri-methyl groups from lysine 4 on histone H3 (H3K4) at transcriptional targets. Affected males typically exhibit language impairment, behavioral disorders, epilepsy, distinctive facial features (small forehead, prognathism, micrognathia, maxillary hypoplasia, facial hypotonia, and a flat philtrum), short stature, or microcephaly (5). In contrast, female carriers of the mutation usually present no symptoms or only mild learning deficits. Although previous cases of KDM5C mutations exist, the unique characteristics of this case highlight its novelty. This data may guide future research on KDM5C gene mutations and assist clinicians in diagnosing different forms of ID. We aim to present a de novo mutation case of XLID due to a novel KDM5C mutation [Xp11.22 (GRCh37)], identified through whole exome sequencing (WES).
2. Case Presentation
2.1. History of Present Illness
A seven-year-old boy presented to the neurology clinic at the Children's Medical Center in Tehran, Iran, with a chief complaint of seizures, restlessness, and speech disturbance. Upon examination, additional clinical manifestations were noted, including global developmental delay, speech difficulties (limited to 3 - 4word phrases), gait problems, a history of tonic-clonic seizures, short stature, refractive errors, and dry eyes. Following the requisition of para-clinic tests, a molecular pathology consultation was requested due to the presence of multiple congenital anomalies.
2.2. Past Medical History
The patient has a history of hypothyroidism and left-hand polydactyly, both of which were corrected by Levothyroxin and polydactyly surgery, respectively. The first episode of seizure occurred at age four, following a fever, and subsequently recurred without fever, presenting as spasms and cyanosis, which were controlled by Levetiracetam. Prior to the seizures, the patient could speak approximately 14 - 15 words, but following the seizures, his vocabulary decreased to 1 - 2 words.
2.3. Physical Examination
The patient's physical features include a coarse face, synophrys, macroglossia, a flat philtrum, a thin upper lip, diastema, and hirsutism (Figure 1).

Figure 1.
The patient's physical features including coarse face, macroglossia, a flat philtrum, a thin upper lip, diastema, and hirsutism
2.4. Imaging and Laboratory Evaluation
Considering the chief complaints of seizures, global developmental delay, and speech difficulties, brain imaging tests (CT, MRI) and electroencephalogram (EEG) were performed to rule out brain structural disorders. The brain computerized tomography (CT) scan was normal, while the magnetic resonance imaging (MRI) revealed evidence of bilateral cerebellar hemispheres atrophy. The EEG showed multifocal epileptic discharge. Due to speech difficulties, an auditory brainstem response (ABR) test was performed to assess hearing thresholds, and the results were unremarkable.
Laboratory results included a normal urine organic acid analysis by gas chromatography–mass spectrometry (GC-MS) and normal amino acid analysis by high-performance liquid chromatography (HPLC) and chromatography. Metabolic screening by tandem mass spectrometry (MS/MS) showed an increased level of C4DC (methylmalonyl carnitine), and a trace amount of mucopolysaccharides (MPS) was found in the urine metabolic test. Elevated levels of C4DC and MPS are associated with multiple metabolic disorders (6). Additionally, the urine reducing substance test was positive, and homocysteine was decreased. Lactate and ammonia levels were normal, while serum TSH was elevated with normal T3 and T4 levels.
Considering the syndromic features of the patient, alongside seizures, abnormal EEG findings, and bilateral cerebellar hemispheres atrophy, coupled with elevated C4DC and trace MPS suggesting a metabolic disorder, a genetic evaluation was performed to rule out various genetic variations.
Genetic consultation revealed unrelated parents with no history of similar features in their families. Given the different clinical and para-clinical symptoms and the probability of multiple congenital anomalies, WES on a 5cc EDTA blood sample was submitted.
2.5. Whole Exome Sequencing
The WES was performed on the Illumina NovaSeq 6000 platform, achieving an exome coverage (on target) of 100X with a read length of 151bp. The results revealed a pathogenic frameshift deletion in the KDM5C gene [rs1569278313, Xp.11.22 (GRCh37); c.807delC exon7/25; Pro269fs] with X-linked recessive inheritance, responsible for Intellectual Developmental Disorder, X-linked syndromic, Claes-Jensen Type (OMIM:300534) (Table 1).
Table 1.
Data Analysis of Whole Exome Sequencing
| Gene Name/ OMIM | Transcript Identifier | SNP Cluster ID | Associated Disease | Inheritance Pattern | DNA Change | Protein Change | Chromosomal Location | Zygosity | ACMG; Classification |
|---|---|---|---|---|---|---|---|---|---|
| KDM5C; #314690 | NM_004187.5 | rs1569278313 | Intellectual developmental disorder, X-linked syndromic, Claes-jensen, Type, [MIM:300534] | X-linked, Recessive | c.807delC, exon7/26 | Pro269fs | Xp.11.22 (GRCh37) | Hemizygous | Pathogenic, (frameshift deletion) |
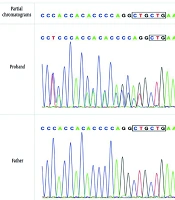
Trio Sanger sequencing was conducted on the patient and his parents to confirm the identified KDM5C mutation. The results verified that the detected KDM5C pathogenic variant in the proband was not present in either parent, indicating the likelihood of a de novo mutation in the proband (Figures 2 -4, Table 1).

Figure 2.
The electropherogram of the patient showed a frameshift deletion in KDM5C: NM_004187.5: Exon7: c.807delC:p.P269fs

Figure 3.
The electropherogram of the patients’ father did not show deletion.

Figure 4.
The electropherogram of the patients’ mother did not show deletion.
2.6. Patient Follow up
Two years after the genetic diagnosis (June 2024), the patient's seizures have been controlled with levetiracetam medication. With speech therapy, he can now say a few words, and occupational therapy has improved his movements and reduced his restlessness. The child's growth has also shown some improvement, although he is developmentally about two years younger than his chronological age. The parents are now considering having a second child.
3. Discussion
Previous reports have demonstrated that KDMs play a crucial role in normal neuronal development and physiological activity (7). Among these, KDM5C encodes a protein with several conserved domains, including the ARID DNA binding domain, the zinc finger domain, the JmjN domain, and two plant homeodomains, all of which are important for chromatin-related processes (4). Research indicates that genetic mutations in the KDM5C gene can adversely affect the levels of H3K4me2/3 modifications in promoter regions responsible for gene expression. Consequently, this can lead to a significant reduction in the levels of two essential proteins, brain-derived neurotrophic factor (BDNF) and voltage-gated channel alpha subunit 2 (SCN2A), which have been linked to both autism spectrum disorder (ASD) and cognitive impairment (8, 9). It is well established that BDNF and SCN2A are crucial for neuronal development, function, and survival (10, 11). Therefore, a deficiency in these proteins may lead to neurological symptoms associated with KDM5C mutations, such as seizures and ID. Additionally, in cases of SCN2A mutation, syndromic facial features like hypertelorism and slanted palpebral fissures may be observed (12).
This study reports on a de novo variant of KDM5C (GRCh37) identified through WES. The mutation was found to be pathogenic based on criteria from the American College of Medical Genetics and Genomics (ACMG), linked to severe ID, seizures, facial dysmorphia, and other previously documented signs and symptoms. This mutation is located in exon 7 of Xp.11.22 and has been submitted three times in the ClinVar database (13), where it has been associated with Claes-Jensen type XLID. One of the ClinVar submissions by Guerra et al. (14) [NM_004187.3(KDM5C):c.807delC; p.(Thr270Glnfs×2)] described three siblings (including male twins and their singleton brother) who shared the same (GRCh37) mutation and exhibited severe ID and speech delay that matched the presenting case report. However, only the oldest brother presented with tonic-clonic seizures. Unlike our case, the twins showed no abnormalities in their MRI scans, and there was no evidence of dysmorphia. Additionally, the carrier mother in their family also displayed the same genetic variation according to Sanger sequencing.
The C nucleotide frameshift deletion observed in the WES of the presenting case aligns with our findings. An investigation on two Taiwanese families with XLID suggested pathogenic novel variations, including c.2233C > G; p.Q745E, which is located on the zinc finger domain, and c.3392_3393delAG; p.E1131Afs, a frameshift variation located near the C terminus of the protein. They noted that mutations in particular functional domains lead to various CNS manifestations. For example, patients with mutations in the zinc finger domains are more prone to severe ID, short stature, and facial abnormalities, while patients with variations near the C terminus more commonly present with autism spectrum disorder (15).
The mentioned study reported four cases of KDM5C mutation, with two cases showing seizures consistent with our findings. Additionally, all individuals exhibited severe ID and speech challenges similar to our case. Our data presented a frameshift deletion on the N terminus in exon 7, which manifests with a syndromic face and short stature, alongside global developmental delay and impaired speech. Another novel KDM5C mutation inherited from the heterozygote proband was c.3344 G > A, which was found to lead to autism and ID in the affected patient (16).
Another instance of a KDM5C mutation was observed in a case from Japan, characterized by severe ID and short stature. The Japanese case exhibited reduced lower extremities and heightened patellar tendon reflexes. Despite these symptoms, the patient was able to walk unaided, unlike our case, which also involved a gait issue. The case study indicated a reduced cerebellar volume on MRI, mirroring the bilateral cerebellar atrophy noted in our research. However, no facial abnormalities were noted in the mentioned case report. Furthermore, the KDM5C mutation in the case report was located in exon 23, contrary to our findings. These differences suggest that while KDM5C mutation may result in shared characteristics, the specific manifestations can vary based on the detailed traits of the mutation (17).
A study explored data on 69 males with KDM5C gene variations and concluded that characteristics like thin lips (0/69) and hypothyroidism (4/69) are rare, as were observed in our present case (5). This could be relevant to the specific mutation in the KDM5C gene identified in this study. The specific findings in this case report, like seizures with abnormal MRI and syndromic facial features, can guide future research to investigate unusual symptoms in KDM5C cases. Additionally, this study helps clinicians to consider KDM5C mutation as a possible differential diagnosis for patients presenting with seizures and developmental delay. Due to the single-patient nature of this case report and the lack of functional validation (cloning in a vector), we cannot draw definite and precise conclusions. A more detailed evaluation of KDM5C cases, with a larger study population, may be substantial. Further investigations are needed to determine the association and mechanisms of action for the effect of KDM5C mutation on clinical manifestations.
Although no treatments are currently available for KDM5C mutation-associated ID, the effects of WNT signaling modulators are being studied (18). The treatment of cancers related to this mutation will also be considered in the future (19). Additionally, genetic counseling is valuable in differentiating fragile X syndrome, which shares many cognitive features with KDM5C mutation-associated ID (18).
In conclusion, this paper presents a case of a novel c.807delC KDM5C mutation and compares the signs and symptoms observed in the patient with those reported in the previous literature. This study provides valuable insight into the pathogenicity and clinical implications of this mutation and may contribute to the understanding and management of potential genetic disorders associated with KDM5C.