





1. Background
Familial Mediterranean fever (FMF) manifests with repeated fever and serous inflammation, including abdominal pain, chest pain, and arthralgia (1). It is mainly prevalent in Turkish, Armenian, North African, Jewish, and Arabic populations (2). It is commonly considered an autosomal recessive disease, and the mutation occurs in the short arm of chromosome 16 in the MEFV gene (3).
This gene encodes the pyrin protein (2), which plays a pivotal role in the innate immunity of the body (4) and detects pathogen activity (5). The mutation of the MEFV gene in these patients, after mild stressors, activates the pyrin protein and subsequently increases its activity, causing inflammation. This leads to the secretion of interleukin 1 (IL-1), IL-18, and other inflammatory mediators, raises chemotaxis, induces neutrophilia, and eventually results in FMF attacks (6).
Although it has diverse clinical manifestations, fever and abdominal pain are the most common symptoms and can be accompanied by peritonitis (7). Secondary amyloidosis is one of its major complications, which has seen a significant reduction with regular colchicine intake (8). Diagnosis is based on clinical symptoms, supported by ethnicity and family history, and confirmed through genetic tests (3).
Colchicine, as the first-line treatment for FMF, is commonly administered to prevent acute attacks, subclinical inflammation, and progressive amyloidosis (9). As noted, FMF has different symptoms and occurs in diverse age groups, races, and genotypes.
2. Objectives
Due to the shortage of evidence on its clinical manifestations and genetic analyses in the Central North of Iran, we aimed to assess the genetic and clinical manifestations of Familial Mediterranean Fever in children.
3. Methods
Referred to the gastroenterology and rheumatology clinics of 17 Shahrivar Hospital, Rasht, Iran from 2011 to 2021. The records of patients with FMF aged < 21 years were included in this study. Familial Mediterranean Fever was diagnosed based on clinical criteria, genetic tests, and response to treatment using the Tel Hashomer method.
3.1. Sample Size
Given the low prevalence of the disease, all records meeting the inclusion criteria were assessed through census sampling. The minimum sample size was determined to be 38, based on the previous study by Twig et al. and the following formula (10):
3.2. Data Gathering
Data were gathered using a checklist that included sex, clinical symptoms, previous abdominal surgery, duration and interval of attacks, family history of FMF, consanguinity of parents, disease severity, type of mutation, drug dose, response to treatment, and complications. Clinical manifestations included fever, abdominal pain, chest pain, arthralgia, erysipelas, hematuria, vomiting, nausea, and diarrhea. Disease complications included amyloidosis, proteinuria, and hematuria.
Disease severity was determined by age at the onset of symptoms, frequency of monthly attacks, presence of arthritis, erysipelas, amyloidosis, and the daily dose of colchicine (Table 1). A higher total score indicated greater severity of the disease. Mild, moderate, and severe disease were noted based on scores of 2 - 5, 6 - 8, and 9 - 19, respectively (11).
| Variables | Scoring |
|---|---|
| Age at the beginning of the symptoms (y) | |
| > 31 | 0 |
| 21 - 30 | 1 |
| 11 - 20 | 2 |
| 6 - 10 | 3 |
| < 6 | 4 |
| The frequency of monthly attacks | |
| < 1 | 1 |
| 1 - 2 | 2 |
| > 2 | 3 |
| Arthritis | |
| Acute arthritis | 2 |
| Chronic arthritis | 3 |
| Erysipelas | 2 |
| Amyloidosis | 3 |
| The daily dose of colchicine | |
| Up to 1 mg | 1 |
| 1.5 mg | 2 |
| 1.5 - 2 mg | 3 |
| > 2 mg | 4 |
Response to treatment was evaluated based on the frequency and severity of attacks before and after colchicine administration. A good response was considered if the treatment caused a relatively complete prevention of the attacks. A moderate response was indicated by a reduced frequency and severity of attacks. However, a weak response was noted if there was no change in the frequency and severity of the disease despite treatment with a high dose (12).
3.3. Genetic Analysis
To define the type of mutation, the FMF strip assay test, which is a complete kit for the detection of twelve mutations in the MEFV gene, was used. This test is based on the reverse-hybridization of biotinylated polymerase chain reaction products. These strips show mutations through an enzymatic color reaction visible to the naked eye. The twelve mutations are E148Q, P369S, F479L, M680I (G/C), M680I (G/A), 1692del, M694V, M694I, K695R, V726A, A744S, and R761H. Homozygote (mutation in similar paired alleles), heterozygote (mutation in one allele), compound heterozygote (mutation in two different alleles), or no mutation were indicated in these twelve genes. In addition, a complex genotype was noted in cases of more than two mutations.
3.4. Ethical Considerations
This study was approved by the ethics committee of the Vice-Chancellor of Research at Guilan University of Medical Sciences (Code: IR.GUMS.REC.1400.515).
3.5. Statistical Analysis
Data were reported as mean, standard deviation, number, and frequency using IBM SPSS Statistics for Windows, Version 26.0 (IBM Corp., Armonk, NY). Qualitative data were analyzed using the chi-square and Fisher’s exact tests. The normality distribution of quantitative data was assessed using the Shapiro-Wilk test. The Independent-T and Mann-Whitney U tests were used to compare quantitative variables. A P-value < 0.05 indicated statistical significance.
4. Results
In this study, 52 records of patients with FMF were assessed. Among them, eight patients did not undergo genetic tests, and there were four incomplete records. Therefore, 12 records were excluded, and we analyzed 40 patients. Among them, 62.5% were boys and 37.5% were girls. Most of the patients were non-consanguineous (80.6%), and 16.7% had a familial history of FMF. The most common clinical manifestations were fever, abdominal pain, nausea, vomiting, chest pain, arthralgia, headache, diarrhea, vertigo, and constipation, respectively. There was no arthritis or erysipelas.
Our results showed that 11.11% of patients had a history of abdominal surgery. Two patients diagnosed with appendicitis underwent appendectomy. Diagnostic laparotomy was performed on one patient with abdominal pain, and one patient had ileum resection due to intestinal obstruction. Additionally, there was one known case of spherocytosis who underwent splenectomy (Table 2).
| Symptoms | No. (%) |
|---|---|
| Fever | 39 (97.50) |
| Abdominal pain | 39 (97.50) |
| Nausea | 19 (50.00) |
| Vomiting | 15 (39.47) |
| Chest pain | 14 (36.48) |
| Arthralgia | 8 (21.05) |
| Headache | 8 (21.05) |
| Diarrhea | 6 (15.78) |
| Limb pain | 4 (10.52) |
| Dizziness | 3 (7.89) |
| Constipation | 1 (2.63) |
| Arthritis | 0 (0) |
| Erythema like erysipelas | 0 (0) |
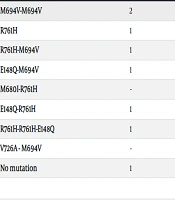
The mean duration of the attacks and the intervals between them in patients were 62.45 ± 47.49 hours and 25.70 ± 33.58 days, respectively. In this study, episodic hematuria and proteinuria were detected in 32.43% and 21.62% of patients, respectively (Table 3). There were no cases of amyloidosis. One patient exhibited swelling, erythema, desquamation, and gastrointestinal symptoms such as diarrhea. Additionally, there was one patient with a probable diagnosis of polyarteritis nodosa who was recommended for abdominal angiography and had no detected mutation.
| Genetic Mutations | Episodic Proteinuria | Episodic Hematuria |
|---|---|---|
| M694V-M694V | 2 | 1 |
| R761H | 1 | 2 |
| R761H-M694V | 1 | 2 |
| E148Q-M694V | 1 | 1 |
| M680I-R761H | - | 1 |
| E148Q-R761H | 1 | 1 |
| R761H-R761H-E148Q | 1 | - |
| V726A - M694V | - | 1 |
| No mutation | 1 | 3 |
In genetic assessments, 12.50%, 47.50%, 20%, 2.50%, and 17.50% of patients were homozygote, compound heterozygote, heterozygote, complex, and no mutation, respectively. M694V/M694V, R761H/M694V, and E148Q/- were the most common homozygote, compound heterozygote, and heterozygote genotypes, respectively (Tables 3 and 4).
| Genetic Mutations | No. (%) |
|---|---|
| Homozygote | 5 (12.50) |
| M694V/M694V | 3 (7.50) |
| R761H/R761H | 1 (2.50) |
| M680I/M680I | 1 (2.50) |
| Compound heterozygote | 19 (47.50) |
| R761H/M694V | 5 (12.50) |
| R761H/M680I | 4 (10.00) |
| M694V/E148Q | 3 (7.50) |
| M694V/M680I | 2 (5.00) |
| R761H/E148Q | 2 (5.00) |
| M694V/V726A | 1 (2.50) |
| R761H/M694I | 1 (2.50) |
| P369S/E148Q | 1 (2.50) |
| Heterozygote | 8 (20.00) |
| E148Q/- | 4 (10.00) |
| M680I/- | 2 (5.00) |
| P369S/- | 1 (2.50) |
| R761H/- | 1 (2.50) |
| Complex | 1 (2.50) |
| R761H/R761H/E148Q | 1 (2.50) |
| No mutation | 7 |
Overall, 59 mutated alleles were found. The most common genetic mutations were R761H (28.2%), M694V (28.2%), E148Q (18.65%), M680I (16.95%), P369S (3.38%), V726A (1.69%), and M694I (1.69%).
The mean severity of disease in patients was 7.12 ± 1.43. The severity of disease was mild, moderate, and severe in 8.83%, 73.53%, and 17.64% of patients, respectively. Among patients, 46.2%, 17.9%, 15.4%, 15.4%, and 5.1% received 1, 2, 1.5, 0.5, and 0.75 mg of colchicine daily, respectively. Most of the patients had good responses to treatment (87.50%), 10% had moderate responses, and 2.50% had weak responses.
Assessing the relationship between clinical symptoms and the most common gene mutations (R761H, M694V, M680I, and E148Q) showed that only chest pain was significantly associated with the R761H and M694V gene mutations (P < 0.05). However, there was no significant relationship between any clinical symptoms and the M680I and E148Q mutations. In addition, results showed a shorter duration of attacks in patients with the R761H mutation and shorter intervals in those with M694V mutations. There was no significant relationship between all four gene mutations and the severity of the disease, the occurrence of hematuria, or episodic proteinuria.
5. Discussion
The genetic assessment of FMF, one of the most common monogenic autoinflammatory diseases in the Middle East, can help clinicians manage patients more accurately. In this study, we found that 12.50% of patients were homozygous, 47.50% were compound heterozygous, 20% were heterozygous, 2.50% had complex genotypes, and 17.50% had no mutation. The most common gene mutations were R761H, M694V, E148Q, and M680I alleles, respectively. The incidence of chest pain was significantly associated with mutations in the R761H and M694V genes.
The sex ratio of male to female was 2:1, consistent with Celikel et al. (13). Although Mattit et al. reported a male to female ratio of 1:4 (14), several other investigations noted no sex preference (12, 15-20).
Our study indicated that 19.4% of parents were consanguineous, which was similar to Rostamizadeh et al. (21). However, Bidari et al. (22) and Celikel et al. (13) reported a higher rate. Additionally, our results showed that 16.7% of patients had a family history of FMF. Despite similar findings by Rafeey et al. (16), other studies reported a higher rate of occurrence (12-14, 21, 22). This emphasizes the potential adverse effect of consanguineous marriage and family history on the risk of FMF.
The mean duration and interval of attacks were 62.45 ± 47.49 hours and 25.70 ± 33.58 days, respectively. These results were consistent with previous Iranian and Turkish investigations. Salehzadeh reported the mean duration of every episode and mean interval between attacks as 36.5 ± 29.6 days and 43.3 ± 34.5 hours, respectively (15). Additionally, Celikel et al. noted three days as the duration and one month as the interval between attacks (13). Therefore, it seems that patients in the Mediterranean region may have consistent patterns in terms of the duration and interval of attacks.
Most of our patients had moderate disease (73.53%), 8.83% had mild disease, and 17.64% had severe disease. While some of our patients had more severe disease, most of them had a favorable response to treatment. Previous investigations reported consistent results (12, 18), emphasizing the importance of proper treatment regardless of disease severity.
In the genetic results, 12.50% of patients were homozygous, 47.50% were compound heterozygous, 20% were heterozygous, 2.50% had complex genotypes, and 17.50% had no mutation. Regarding the type of genes, although there are Turkish, Iranian, Egyptian, and Lebanese studies that mentioned a higher frequency of no mutation (12, 16, 17, 19-21), Bidari et al. (Iranian) (22) and Hageman et al. (Dutch) (18) found a lower frequency. There are also consistent investigations by Celikel et al. (Turkish) (13) and Mattit et al. (Syrian) (14). Moreover, we found that R761H, M694V, E148Q, M680I, P369S, V726A, and M694I were the most common genetic mutations. Salehzadeh (15) and Mattit et al. (14) found M694V and V726A; Bidari et al. (22), Ozalkaya et al. (12), and Hageman et al. (18) noticed M694V and M680I; Cekin et al. (17) and Rostamizadeh et al. (21) reported M694V and E148Q; Mansour et al. (19) and El Roz et al. (20) demonstrated E148Q and V726A as the most common involved alleles. Furthermore, our patients with the mutation in the R761H gene had the lowest age at the beginning of symptoms; however, Celikel et al. (13) found an M694V mutation. These discrepancies indicate that FMF in different regions follows different patterns and there is a need for thorough genetic assessment in each patient.
In the present study, there was no significant relationship between fever and abdominal pain with any of the mutations. Although there are Iranian and Turkish investigations (15-17, 21) that noticed M694V mutations in patients with fever and abdominal pain, Mansour et al. (19) found the E148Q mutation in these patients. Our results showed that chest pain had a significant relation with the R761H and M694V genes. Consistently, Salehzadeh (15) found M694V, and Cekin et al. (17) noticed M680I and M694V mutations in patients with chest pain. These similar results indicated that M694V should be noted in patients with chest pain presentation.
In our study, there was no significant relation between the mutations in genes and the severity of the disease or the responsiveness to treatment. However, Cekin et al. (17) found more severe symptoms in patients with M694V and M680I and milder symptoms in E148Q and V726A. Besides, Rostamizadeh et al. (21) found the M694V mutation in patients with more severe symptoms.
5.1. Limitations
Although we successfully assessed the clinical and genetic results of our patients who were referred to our tertiary referral medical center, this study had some limitations. We used the FMF strip assay test, which can evaluate twelve mutations. It would be more informative if we could assess our patients with advanced methods such as whole exome sequencing. Additionally, given the single-center nature of our study and the limited sample size, further multicenter investigations with larger sample sizes and more advanced methods are recommended.
5.2. Conclusions
This study indicated that FMF in different regions follows different patterns and there is a need for thorough genetic and clinical assessments in each patient. Determining the clinical and genetic results can help clinicians detect patients rapidly, inhibit unnecessary diagnostic and therapeutic processes, and prevent long-term complications. Additionally, based on the recessive autosomal hereditary pattern and the presence of clinical manifestations in patients without mutations or in carriers of only one mutation, it seems that other genetic factors, in addition to mutations in MEFV, can affect FMF morbidity. Therefore, we recommend further investigations into the environmental and genetic factors affecting patients.
