


1. Introduction
Vici syndrome (OMIM #242840) is a rare, autosomal recessive progressive disorder characterized by agenesis of the corpus callosum, skin and hair hypopigmentation, cataracts, variable immunodeficiency, cardiomyopathy, and severe developmental delay. It was first described in a sister and brother in 1988 (1). The syndrome is associated with homozygous or compound heterozygous mutations in the ectopic P-granule 5 (EPG5) gene located on chromosome 18q (2). Autosomal recessive variants in EPG5, an essential autophagy regulator, are known to be the genetic cause of this syndrome (3, 4). Several features of the disease, such as agenesis of the corpus callosum and skin hypopigmentation, are present at birth, while other pathologies, including cardiomyopathy and immunodeficiency, may emerge and progress later in life (1-5).
The clinical significance of identifying a novel mutation is crucial for determining disease risk, progression, response to treatment, and genetic risks for other family members. A phenotypically driven genetic analysis, focusing on genes associated with specific clinical findings and identified variants, strengthens genetic databases and may clarify additional phenotypic findings in newborns. Identifying such variants enhances our understanding and interpretation of complex biological processes.
The discovery of a new mutation is vital for genetic counseling and prenatal testing in future pregnancies. Clinical decisions can be made more precisely and effectively using patient-specific genetic information (6), which offers significant advantages for personalizing the patient’s treatment process and improving outcomes.
We present a 2-month-old girl with Vici syndrome, found to have a homozygous c.7504delC (p.Gln2502Argfs*4) frameshift variant through EPG5 gene sequence analysis.
2. Case Presentation
A two-month-old Turkish girl was admitted to our pediatric neurology outpatient clinic with failure to gain weight and growth retardation. During follow-up, she was found to be restless, crying constantly, and unable to gain weight. She had a birth weight of 3450 g at term, a head circumference of 32 cm, and was delivered by spontaneous vaginal delivery. Her family history included second-degree consanguinity between her parents.
On neurological examination, the patient presented with microcephaly, a high palate, an open mouth with an inverted lip appearance, hypopigmented hair and skin, diffuse hypotonia, hyperlaxity, and weak developmental reflexes, while deep tendon reflexes were normal (Figure 1).

Nasogastric tube inserted, high palate, mouth open and inverted v-lip view, there is hypotonicity, head back in traction, hypopigmented skin appearance
Other systemic examinations revealed normal eye movements, bilateral anterior polar cataracts in the anterior segment examination, slightly hypopigmented retina, normal optic disc appearance, and no nystagmus. Hearing was normal. Cardiac examination revealed a murmur, and echocardiography showed hypertrophic cardiomyopathy and mitral regurgitation.
Given that her clinical signs and symptoms were common to various neurological, metabolic, and neuromuscular diseases, laboratory and imaging tests were performed before genetic diagnostic analysis for differential diagnosis and treatment planning. Laboratory findings included a complete blood count, thyroid function tests, and vitamin B12 and vitamin D levels, all within normal limits. Serum biochemistry showed elevated levels with CK: 828 U/L (29 - 200), AST: 137 U/L (5 - 40), and ALT: 97 U/L (5 - 40). Immunoglobulin levels and screening for TORCH, EBV, and parvovirus infections were normal, which helped assess the etiology of the elevated liver enzymes. Parameters for celiac disease, carnitine and acylcarnitine levels, urine organic acid analysis, and biotinidase activity were also within normal ranges in the assessment for neuromotor developmental delay.
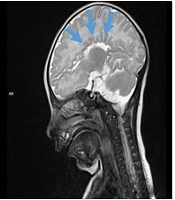
Magnetic resonance imaging (MRI) of the brain revealed agenesis of the corpus callosum, mild dilation of the third ventricle, and colpocephalic dilation of the lateral ventricle (Figure 2). Based on these laboratory and imaging results, congenital infectious diseases and metabolic disorders were excluded in our patient, who presented with growth retardation, hypotonia, cataracts, and elevated liver enzymes. Following a thorough physical examination by our geneticist, it was decided to proceed with targeted genetic testing for neurogenetic diseases. Gene analysis for spinal muscular atrophy and Pompe disease, relevant to hypotonic infant etiology, was normal.
, agenesis of the corpus callosum (blue arrows), on axial T1, sagittal T2 and FLAIR sequences.")
Examined brain MRI findings; mild dilatation in the third ventricle and colpocephalic dilatation in the lateral ventricle (red arrows), agenesis of the corpus callosum (blue arrows), on axial T1, sagittal T2 and FLAIR sequences.
The EPG5 gene sequence analysis was subsequently planned, considering Vici syndrome as a preliminary diagnosis based on the presence of hypertrophic cardiomyopathy, bilateral congenital cataracts, hypotonia, agenesis of the corpus callosum, and hypopigmented appearance. Sanger sequence analysis revealed a homozygous c.7504delC (p.Gln2502Argfs*4) frameshift variant. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, this variant was classified as likely pathogenic. Its pathogenicity was evaluated using various online analysis tools, including MutationTaster, SIFT, VarSome, and PolyPhen-2, all of which suggested a high likelihood of disease causation. This variant was not reported in the 1000 Genomes Project, the Exome Aggregation Consortium (ExAC) Browser, gnomAD, or the Human Gene Mutation Database (HGMD). The 1 bp deletion in the EPG5 gene is predicted to be functionally deleterious, resulting in a frameshift that causes premature termination of the protein and leads to nonsense-mediated decay (NMD).
Parental screening identified both parents as heterozygous carriers of the variant. Since there is no known cure for Vici syndrome, supportive treatment was provided. Our patient was fed via a nasogastric catheter and was under follow-up care from pediatric cardiology, physiotherapy, and ophthalmology departments. Genetic counseling was recommended. However, due to irregular follow-up visits, long-term outcomes could not be observed.
3. Discussion
In most reported cases, this syndrome is associated with several typical features, including developmental delay (97.4%), recurrent infections (98.73%), agenesis of the corpus callosum (97.4%), immunodeficiency (75.94%), intellectual disability (80 - 99%), cataracts (75%), cardiomyopathy (65%), seizures (65%), and renal abnormalities (15%) (4,7,8). Vici syndrome can be considered a significant cause of developmental delay (7). In our case, a patient presenting to our outpatient clinic with growth retardation was diagnosed early by targeting the gene based on examination findings.
Vici syndrome is recognized as a progressive neurodevelopmental multisystem disorder (4, 8). Elevated liver enzymes have been noted in the literature (9, 10), and moderate elevation in liver enzymes was also observed in our patient. Although congenital cataract is one of the classical diagnostic features of Vici syndrome, other ocular findings such as optic neuropathy, nystagmus, and mild ptosis have also been reported (11). In our case, bilateral anterior polar cataracts were present in the anterior segment examination. The retina was slightly hypopigmented, with a normal optic disc appearance and no nystagmus.
While seizures are common in this syndrome, our patient had no history of seizure activity.
Our patient presented with hypotonia, hypertrophic cardiomyopathy, bilateral congenital cataracts, agenesis of the corpus callosum, and hypopigmented skin, findings also reported in the literature (4, 12-18). Based on these findings, a novel homozygous variant, c.7504delC (p.Gln2502Argfs*4), resulting in a frameshift mutation, was identified. Cardiomyopathy is one of the most common causes of death in patients with Vici syndrome, with cases of rapid progression to heart failure documented (11). The early presence of cardiomyopathy in our patient suggests a poor prognosis.
In recent years, the clinical spectrum of Vici syndrome has broadened with reports of additional findings, such as laryngomalacia, pharyngomalacia, idiopathic thrombocytopenic purpura, and bilateral sensorineural hearing loss (19, 20). However, our patient did not exhibit hearing loss, and both her respiratory pattern and hematological laboratory values were normal. Although mutation types are unspecified in some cases , the disease-causing mutations have been identified in many cases (4, 12-17).
The EPG5 gene contains 44 exons, with its longest transcript encoding a protein of 2,579 amino acids. Cullup et al. identified 20 distinct mutations in the EPG5 gene among 18 patients exhibiting clinical signs of Vici syndrome. Biallelic mutations in EPG5 have been confirmed in over 90% of patients previously diagnosed with Vici syndrome, with locus heterogeneity detected in only a very small subset of cases (4). The mutations identified by Cullup spanned nearly the entire gene, ranging from exons 2 to 39.
A severe defect in autophagosomal clearance linked to mutations in EPG5 was found to cause autophagic cargo accumulation and impaired fusion with lysosomes. Autophagy is essential for cellular homeostasis, facilitating the degradation and recycling of damaged organelles, misfolded proteins, and other cellular debris. Disruption in autophagy significantly impacts multiple tissues, accounting for the multisystemic nature of Vici syndrome. For example, defective autophagy in neurons can lead to the buildup of toxic proteins and damaged organelles, contributing to neurodevelopmental abnormalities like agenesis of the corpus callosum (4, 18).
Defects in autophagy are also linked to cardiomyopathy, neurodegeneration, immune dysfunction, pigmentation defects, oncogenesis, and impaired embryonic development. However, the specific role of defective autophagy in the development of congenital cataracts remains unexplained (4).
According to the study by Byrne et al., most truncated EPG5 mutations were associated with a severe phenotype. However, the only recurrent missense mutation in their series, p.Gln336Arg, was linked to a lower likelihood of cataracts or cardiomyopathy and a relatively longer life expectancy. They also found that patients with compound heterozygous EPG5 mutations tended to have a longer lifespan compared to those with homozygous EPG5 mutations (5). Our patient, diagnosed at 2 months of age through targeted gene testing, could not be evaluated for long-term outcomes as the family discontinued follow-up after receiving the genetic diagnosis.
In a case study by Ehmke et al., gene analysis of a patient with typical clinical symptoms revealed that the mutation in EPG5 was located in exon 43 (the penultimate exon), likely resulting in NMD (13). Another case involved a missense mutation in EPG5 (c.3389A > C) and a microduplication in the exon 1 region, reported in a 3-year-old Japanese girl with Vici syndrome who presented with persistent diarrhea (21).
We analyzed some cases with rare EPG5 mutations reported over the past decade according to their phenotype-genotype characteristics and compared them with our case, as summarized in Table 1. The genetic analysis results in this case, when considered alongside variants previously reported in a few patients, suggest that truncating mutations may play a significant role in the mechanism of the disease.
| Previously Reported Cases | Mahjoubi et al. 2022, (7) | Vansenne et al. 2022, (14) | Abidi et al. 2020, (11) | Moirangthem et al. 2019, (15) | Alzahrani et al. 2018, (16) | Hedberg-Oldfors et al. 2017, (10) | Taşdemir et al. 2016, (9) | El-Kersh et al. 2015, (17) | Our Case |
|---|---|---|---|---|---|---|---|---|---|
| Number of patients | 1 | 15 | 1 | 1 | 1 | 1 | 2 | 1 | 1 |
| Age | 5 years | Range 13 months–17 years | 6 months | 8 months | 5 months | 1 months | 3 months, Birth | 5 years | 2 months |
| Gender | F | M: 6 F: 9 | F | M | M | M | M:2 | F:1 | F |
| Ethnicity | Iranian | NA | Saudi | NA | Saudi | Arabian | Turkish (2) | NA | Turkish |
| Parental consanguinity | (+) | NA | (+) | (+) | (+) | (+) | (+) (2) | (+) | (+) |
| Beginning age | NA | First year of life 4/9 After first year of life 5/9 | Birth | NA | Birth | Birth | NA, After the birth | NA | After the birth |
| Eye findings | Optic atrophy, blindness | 15/15 abnormal vision 11/14 optic hypoplasia 2/14 optic atrophy | Bilateral congenital cataract | Bilateral congenital cataract | Bilateral congenital cataract | Bilateral congenital cataract | Bilateral congenital cataract (2) Flair retina | Optic atrophy, bilateral congenital catract | Bilateral congenital cataract, hipopigment retina |
| Developmental delay | (+) | (+) 15/15 (profound) | (+) | (+) | (+) | (+) | (+) (2) | (+) | (+) |
| Hypotonia | NA | 15/15 | (+) | (+) | (+) | (+) | (+) (2) | (+) | (+) |
| Seizure | (+) | 15/15 | (+) | (+) | (-) | (-) | (-) (2) | (+) | (-) |
| Immunodeficiency | NA | NA | (-) | NA | (+) | (+) | (-) (2) | (+) | (-) |
| Heart findings | NA | (-) | (+) | (+) | (+) | (+) | (+) (2) | (+) | (+) |
| Reflexs | NA | 10/14 brisk | NA | Absent | NA | Absent | NA (2) | NA | (+) |
| Spasticity | (+) | 9/15 | (-) | (+) | NA | NA | NA (2) | NA | (-) |
| Skin findings | NA | (-) | (-) | Mild hypopigmentation | Hypopigmentation | Light pigmentation | Fair hair and skin (2) | Generalized hypopigmentation | Hypopigmentation |
| Corpus callosum | Agenesis | 15/15 agenesis | Agenesis | Agenesis | Agenesis | Agenesis | Agenesis (2) | Agenesis | Agenesis |
| Genetics | EPG5 novel homozygous nonsynonymous mutation | All patients EPG5 missense novel homozygous missense mutation | EPG5 homozygous mutation on exon 27 | EPG5 homozygous novel mutation | EPG5 | Novel homozygous one-base deletion in EPG5 | EPG5 homozygous mutation (2) | EPG5 homozygous missense mutation | EPG5 homozygous frameshift variant |
| Protein change | p.Y1069C Het | p.Arg1621Gln | p. (Leu1584*) | p.Glu1182* | p (Gln1231Gln) | p.I262Sfs*15 | p.R2483* (2) | p.Gln336Arg | p.Gln2502Argfs*4 |
| Nucleotide change | c.A3206G | c.4862G > A | c.4751T>A | c.3544G>T | c.3693G>A | c.784delA | c.7447C > T (2) | c.1007A>G | c.7504delC |
| ACMG classification | VUS | P | P | P | VUS | LP | LP | P | P |
| Creatine kinase | NA | NA | N | 358 U/L | NA | 765 U/L | 213–491 U/L, 1563 U/L | NA | 828 U/L |
The Review of Cases with the Rare Ectopic P-granules Autophagy Protein 5 (EPG5) Mutation Based on Their Phenotype-genotype Characteristics a
In our case, a 1 bp deletion in the EPG5 gene was predicted to be functionally deleterious, causing a frameshift and premature termination of the protein, resulting in NMD. Notably, optic atrophy and nystagmus, which are clinically expected findings in Vici syndrome, were absent in our patient. However, our patient did present with bilateral anterior polar cataracts. In children with Vici syndrome, cataracts typically tend to be visually insignificant. In this case, ocular findings were not severe, but hypertrophic cardiomyopathy—a known life-threatening feature—was present early on with this variant.
Currently, there is no specific treatment for Vici syndrome; therapeutic interventions are mainly supportive, aiming to prolong survival and alleviate symptoms. Upon diagnosing Vici syndrome in our patient, we provided the family with genetic counseling regarding recurrence risks. Prenatal genetic diagnosis options were explained, including the possibility of preimplantation genetic diagnosis. It was emphasized that consanguineous marriage is a significant risk factor in this rare genetic disorder.
Identifying novel variants holds important implications, as they may eventually guide targeted treatment options. Potential treatment strategies under discussion for this complex disease include therapeutic approaches targeting the autophagy pathway, such as enhancing autophagic activity, gene therapy, and modulation of downstream effects (22).
We hope that presenting new cases and gaining a better understanding of the autophagy pathway in more patients will support the future development of targeted therapies.
A limitation of this case report is the lack of long-term follow-up and additional genetic testing in family members. Future research should aim to further investigate the impact of the newly identified variant on Vici syndrome, potentially through long-term follow-up studies or additional genetic testing in larger cohorts.
In conclusion, a new variant was identified in a patient diagnosed with Vici syndrome through targeted genetic analysis. Examining genes known to be associated with clinical findings and identified variants strengthens genetic databases and may help clarify additional phenotypic findings in newborns, if present. Moreover, knowledge of such variants is critical for genetic counseling and prenatal testing in future pregnancies.
