




1. Introduction
Polyarteritis nodosa (PAN) is a necrotizing vasculitis affecting small-to-medium-sized arteries without the involvement of glomerulonephritis or antineutrophil cytoplasmic antibody (ANCA) (1). The PAN has two major subtypes: Systemic PAN and cutaneous polyarteritis nodosa (cPAN). While systemic PAN frequently involves multiple organs, including the kidneys, joints, muscles, nerves, and gastrointestinal tract, cPAN is restricted to the skin and subcutaneous tissue. Interestingly, 28% to 60% of patients with systemic PAN also present with cutaneous findings such as palpable purpura, livedo reticularis, and subcutaneous nodules (2). However, cPAN, which is considered a distinct single-organ vasculitis, predominantly affects the dermis-subcutis junction and the subcutis itself, with a typically chronic but favorable course (2).
Although cPAN can occur at any age, from infancy to late adulthood, with an average age of onset around 40, it is rarely encountered in the pediatric population (2). Due to the lack of universally accepted diagnostic criteria for cPAN, it is typically diagnosed based on clinical presentation and histopathological findings. The hallmark histopathologic feature of cPAN is segmental necrotizing vasculitis of medium- and small-sized arteries or arterioles (2-4). Other characteristic skin lesions include subcutaneous nodules, livedo reticularis, purpura, and ulcers. There are also several exclusion manifestations, such as long-lasting fever, weight loss, hypertension, and rapidly progressive renal failure (2).
Given that cPAN may present with overlapping features of systemic PAN and other vasculitic syndromes, differential diagnosis is often challenging, requiring extensive workup (3). In parallel with its rarity in the pediatric population, little is known about the full spectrum of clinical presentations and optimal management strategies for pediatric cPAN.
In view of the foregoing, we conducted this case study featuring a 5-year-old female cPAN case after obtaining written consent from her parents and paying due attention to the principles outlined in the Declaration of Helsinki, explaining in detail the clinical manifestations, diagnostic findings, and treatment approach.
2. Case Presentation
The case featured in this case report is a 5-year-old girl presented to the pediatric outpatient clinic with complaints of rashes that first appeared on the dorsal sides of her feet and then on the dorsum of her hands. Her current height and weight were measured at 125 cm and 28 kg, respectively, within normal percentiles. She had no known comorbidities. She had an upper respiratory tract infection one week before the onset of the rashes.
She was born at 38 weeks of gestation via spontaneous vaginal delivery as the fifth child in a family with six healthy siblings. Her parents were maternal cousins. Her household had no pets, and she had not traveled in the past three months.
Following hospitalization, she was started on oral amoxicillin-clavulanate and discharged without complications. However, two months later, she developed polymorphic painful rashes.
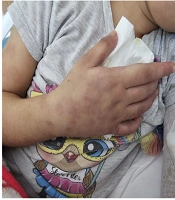
Her physical examination revealed painful skin lesions predominantly on the hands' dorsum and distal lower extremities, including the ankles, dorsum of the feet, heels, and toes. In addition, palpable, mobile, and non-indurated cervical lymphadenopathy measuring less than 2 cm was noted on both sides of the neck. The vascular access on the back of the right hand could not be thoroughly evaluated. Nonetheless, there was edema in both feet. A netlike pattern of reddish-blue skin discoloration (livedo reticularis) characterized by a reticular, violaceous pattern was present on the lower limbs (Figures 1 - 3).

Frontal view of the patient showing no facial deformities but depicting overall well-being at presentation. This image was taken during hospitalization and is included to document general appearance prior to systemic treatment initiation.

Livedo-like violaceous discoloration on the dorsum of the hand and forearm. These lesions were painful to touch and one of the initial cutaneous signs prompting further diagnostic workup. The image also shows soft tissue swelling and subcutaneous discoloration.
 observed on the dorsum and lateral aspects of both feet, accompanied by mild edema. These findings are consistent with medium-vessel vasculitis localized to the dermo-subcutaneous region, characteristic of cutaneous polyarteritis nodosa (cPAN).")
Reticular, violaceous skin lesions (livedo reticularis) observed on the dorsum and lateral aspects of both feet, accompanied by mild edema. These findings are consistent with medium-vessel vasculitis localized to the dermo-subcutaneous region, characteristic of cutaneous polyarteritis nodosa (cPAN).
The results of the laboratory tests are summarized in Table 1. The result of the autoinflammatory disorders gene panel test came out negative, indicating the absence of mutations. Bone marrow analysis, urine culture, and thorax computed tomography (CT) findings did not reveal any abnormality. The result of the deficiency of adenosine deaminase 2 (ADA2) genetic test was also negative.
| Parameters | Value |
|---|---|
| Hemoglobin (g/dL) | 8.4 |
| White blood cell count (× 103/L) | 30.790 |
| Neutrophil count (× 103/L) | 23.790 |
| Platelet count (× 103/L) | 488.000 |
| Aspartate aminotransferase (IU/L) | 210 |
| Alanine aminotransferase (IU/L) | 105 |
| Lactae dehydrogenase (IU/L) | 382 |
| Antistreptolysin O (units/mL) | 4.094 |
| Erythrocyte sedimentation rate (mL/h) | 100 |
| C-reactive protein (mg/dL) | 220 |
Laboratory Investigations of the Child
A skin punch biopsy was performed on one of the cutaneous lesions on the left arm. Histopathological examination of the biopsied specimen revealed orthokeratosis in the epidermis, regular epithelial structure, sparse perivascular lymphocytes in the upper dermis, neutrophil-rich inflammatory cells in the walls of medium-sized vessels in the subcutaneous fat tissue, fibrinoid necrosis, scattered nuclear fragments, rare eosinophils, and prominent neutrophils. Periodic acid-Schiff-alcian blue staining revealed fungal structures on the surface, whereas Ehrlich-Ziehl-Neelsen staining did not reveal any acid-fast bacilli.
The clinical findings and histopathological evidence indicating medium-sized vasculitis suggested the preliminary diagnosis of cPAN, while the diagnosis of DADA2 was excluded due to the lack of relevant mutation. Accordingly, the patient was started on oral prednisone at a dose of 2 mg/kg per day along with 200 mg/day prophylactic isoniazid due to positive (10 mm of swelling or more) purified protein derivative (PPD) and QuantiFERON test results. The absence of parenchymal infiltrations on thoracic CT, three negative polymerase chain reaction (PCR) test results of gastric fasting fluid, the absence of acid-resistant bacteria, and negative mycobacterial antigen immunohistochemistry test result indicating no fungal culture led us to rule out mycobacterial infection. In the second week, the patient was started on 2.5 mg/kg/day azathioprine and colchicine as her rashes did not resolve. She was given intramuscular benzylpenicillin over the following three months at 21-day intervals. Prednisone treatment was tapered and subsequently discontinued by the sixth month of treatment when the rashes resolved entirely. The patient continued to be symptom-free at the one-year follow-up visit while she was on azathioprine, colchicine, and isoniazid.
3. Discussion
This case report features a child diagnosed with cPAN based on clinical and histological findings. As a rare form of pediatric vasculitis, approximately 200 cases of cPAN have been reported in the literature since Kussmaul and Maier first described it in 1866 (5). Our patient, who exemplified the typical presentation of cPAN, underscores the importance of recognizing this condition to ensure timely diagnosis and appropriate management.
The estimated prevalence of PAN is approximately 31 cases per million, with cPAN accounting for about 4% of all PAN cases (6, 7). Historically, pediatric PAN has been categorized into three subtypes: Systemic PAN, cutaneous PAN, and infantile PAN. However, since it was later determined that infantile PAN, in fact, represents a severe variant of Kawasaki disease, pediatric PAN is now categorized into two subtypes: Systemic PAN and cutaneous PAN. First described as a distinct entity in 1931, cPAN is characterized by necrotizing vasculitis involving medium- and small-sized vessels of the dermis-subcutis junction and the subcutis itself (8).
Several clinical features help distinguish cPAN from systemic PAN. In cPAN, vasculitis is restricted to the skin, whereas systemic PAN often involves multiple organ systems, including the kidneys, joints, muscles, peripheral nerves, and gastrointestinal tract. Although cPAN is limited to the skin, extracutaneous involvement featuring the muscles and nerves under the skin has been reported in more than 60% of the affected cases (9, 10).
The differential diagnosis of pediatric patients with vasculitic skin lesions is often challenging. The diseases that should be considered in the differential diagnosis of children with cutaneous lesions suggestive of vasculitis, including immunoglobulin A (IgA) vasculitis (Henoch-Schönlein purpura), Kawasaki disease, childhood PAN, and ANCA-associated vasculitis (11), are listed in Table 2. Additionally, disorders such as hepatitis B virus (HBV)-associated necrotizing vasculitis and DADA2 can mimic PAN-like manifestations (12). Therefore, a comprehensive evaluation, including clinical, histopathological, and immunological testing, is required for an accurate differential diagnosis (11, 13).
| Disease | Size of Vessel Involved | Involved Organs | Cutaneous Manifestations | Coexisting Symptoms | Histopathology |
|---|---|---|---|---|---|
| IgA vasculitis | Small-sized | Skin, synovial membrane, gastrointestinal tract, and kidneys | Non-thrombocytopenic purpura or petechia, primarily on the lower limbs | Nonerosive arthritis and arthralgia | Neutrophilic infiltration and immune complex deposits, primarily involving IgA, in the walls of arterioles, capillaries, and venules |
| ANCA-associated vasculitis | Small to medium-sized | Kidneys, upper airway, lungs, and skin | Palpable purpura, hemorrhagic blisters, tender subcutaneous nodules, livedo reticularis/racemosa, painful ulcers mimicking pyoderma gangrenosum, and digital gangrenes | Fever, fatigue, weight loss | Necrotizing vasculitis, granulomatosis with polyangiitis |
| Kawasaki disease | Medium-sized | Coronary arteries, conjunctiva, lips, oral cavity, and neck | Erythema, edema, desquamation, polymorphous exanthema | Fever, conjunctivitis, cracked lips with erythematous oropharynx, rash, lymphadenopathy, erythema of palms, and soles | Necrotizing vasculitis with fibrinoid necrosis of the coronary arteries |
| Systemic polyarteritis nodosa | Medium- to small-sized | Skin, musculoskeletal, renal, gastrointestinal, and central nervous system | Palpable purpura, livedo reticularis, and subcutaneous nodules | Fever, myalgia, arthralgia/arthritis, renal involvement, and neurologic involvement | Fibrinoid vascular necrosis of the vessel wall with a mixed infiltrate consisting of various combinations of leukocytes and lymphocytes |
| Deficiency of ADA2 | Small to medium-sized | Systemic vasculitis, early-onset stroke, bone marrow failure, and/or immunodeficiency | livedoid and non-specific nodular rashes (livedo racemosa/reticularis), subcutaneous nodules and macular erythema | Hypertension, headache, fatigue, myalgia/arthralgia, arthritis, myositis and intermittent fever | Nongranulomatous, necrotizing arteritis of a medium-sized vessel resembling cPAN in some cases, thrombosis of dermal and/or hypodermal capillaries, without vasculitis |
Differential Diagnosis of Children with Cutaneous Lesions, Suggestive of Vasculitis
The 1990 American College of Rheumatology (ACR) diagnostic criteria for PAN suggest that patients with one cutaneous and one extracutaneous feature can be diagnosed with PAN, provided that histopathological findings also support PAN diagnosis (14). Although specific diagnostic criteria for cPAN have been proposed by a Japanese research group (10), they have not yet been universally accepted. The characteristic histopathologic feature of cPAN is leukocytoclastic vasculitis with fibrinoid necrosis in small-to-medium-sized arteries in the deep dermis and/or panniculus, while its other features include inflammatory nodules, necrotizing arteritis with inflammatory infiltrates, leukocytoclasia, edema, and immunoglobulin M (IgM) and complement component 3 (C3) deposition in vessel walls (15, 16). Taken together, these findings indicate that detecting diagnostic histological findings in patients with consistent clinical signs and symptoms may be the gold standard for diagnosing PAN, particularly cPAN.
Recent studies have identified biallelic mutations in the ADA2 gene in a subset of pediatric PAN cases, particularly those with early disease onset or a positive familial history (17). In parallel, DADA2 was found to be associated with cutaneous vasculitis, systemic inflammation, variable hypogammaglobulinemia, and early-onset ischemic strokes (12, 18). Our patient's DADA2 genetic analysis did not reveal pathogenic mutations. Given its clinical overlap with PAN, DADA2 should be suspected in children presenting with early-onset strokes, immunodeficiency, or bone marrow dysfunction.
The prognosis for cPAN is generally favorable, with a low likelihood of progression to systemic PAN (19). However, its chronic and relapsing nature poses therapeutic challenges. Therefore, cPAN treatment decisions are typically made based on the severity of the disease (20, 21). Accordingly, while patients with mild disease, characterized by normal renal functions and absence of end-organ damage, are managed with corticosteroids and colchicine (18, 22, 23), those with relapsing disease are managed with steroid-sparing agents such as azathioprine, methotrexate, or mycophenolate mofetil. Although intravenous immunoglobulin (IVIG), tumor necrosis factor-α (TNF-α) inhibitors, and interleukin-6 (IL-6) antagonists have been used in refractory cases, their efficacy has yet to be established (11, 13).
Our patient was treated with prednisone, colchicine, and azathioprine as primary treatment and isoniazid as prophylactic treatment. She was discharged after one month of hospitalization, with her prednisone treatment tapered off. At the one-month follow-up visit, she was afebrile and asymptomatic, and her cutaneous lesions had resolved.
Pediatricians should consider cPAN in children presenting with persistent cutaneous vasculitis, especially in those with recurrent tender subcutaneous nodules, livedo reticularis, or ulcerations in the absence of systemic organ involvement. Given the overlapping clinical features of cPAN, systemic PAN, and other vasculitides, early histopathological confirmation via skin biopsy is crucial to establish an accurate diagnosis and avoid unnecessary delays in treatment.
Although corticosteroids and colchicine continue to be the mainstay of cPAN treatment, emerging therapies, including biologics, may be considered as alternative treatment options in refractory cases. Further longitudinal studies are needed to better characterize long-term disease progression, optimize treatment strategies, and establish standardized management guidelines for pediatric cPAN.
This case report had several limitations. First, the fact that it is a single-patient observation does not allow its findings to be generalized to the broader pediatric cPAN population. Secondly, the lack of extended follow-up beyond the initial treatment period limits our ability to assess long-term relapse risk and treatment efficacy. Lastly, while histopathological findings confirmed the diagnosis, additional immunological and genetic analyses, such as ADA2 mutation screening in a larger cohort, may have provided further insights into disease pathogenesis and potential therapeutic targets. Future multicenter studies or prospective cohort analyses are needed to define the optimal treatment approach and long-term outcomes for pediatric cPAN.
In conclusion, given its clinical overlap with systemic PAN and other vasculitides, early accurate diagnosis through histopathological confirmation with skin biopsy is critical for timely treatment of cPAN, a rare pediatric vasculitis presenting with persistent cutaneous lesions including subcutaneous nodules, livedo reticularis, and ulcerations. The cPAN generally follows a chronic but benign course. However, long-term clinical follow-up is essential as relapses may occasionally occur. Corticosteroids and colchicine remain the first-line treatments for cPAN, while immunosuppressive agents may also be used in refractory cases. Emerging therapies such as IVIG and biologics may be considered alternative treatment options in severe or relapsing cases. There is a need to increase awareness of cPAN among clinicians and conduct further research to develop standardized treatment protocols for pediatric cPAN and improve its long-term outcomes.
